Drug fights cystic fibrosis
An experimental drug that has proven effective in treating muscular dystrophy also works for cystic fibrosis, according to researchers at the University of Alabama at Birmingham (UAB).
The new study is the latest on a compound called PTC124, which helps to “rescue” faulty proteins that lead to illnesses. The drug holds promise in treating more than 2,400 genetic diseases caused by a certain class of DNA mutation.
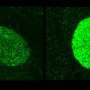
In the UAB tests performed on mice, PTC124 restored to normal function up to 29 percent of the cases of abnormal cystic-fibrosis (CF) protein.
The findings are published in the online version of the journal Proceedings of the National Academy of Sciences and will soon appear in a print edition.
The study adds to research published last year in the journal Nature, which showed PTC124 restored up to 25 percent of the missing or abnormal protein function in mice with Duchenne muscular dystrophy.
“Our study shows strong pre-clinical evidence that PTC124 is capable of suppressing ‘nonsense mutations’ that cause cystic fibrosis,” said David Bedwell, Ph.D., professor in the UAB Department of Microbiology and lead author on the study. “We think this provides strong evidence for clinical trials with PTC124 in CF patients with this kind of mutation.”
A gene that carries a ‘nonsense mutation’ produces a shortened or faulty protein that degrades in the body. The absence of that protein is what leads to disease, Bedwell said. An estimated one-third of gene defects responsible for human disease are thought to come from ‘nonsense mutations.’ In the case of CF, the absence of a certain protein leads to an imbalance of salt and water in the linings of the lungs and other membranes.
The UAB study showed that PTC124 allowed the protein to be made in mouse cells where it was previously absent, and it was delivered in a specific location that helped restore salt and water balance in membranes.
“The preclinical and clinical data on PTC124 support our hope that this drug will be an important disease-modifying therapy for cystic fibrosis,” said Robert J. Beall, Ph.D., president and chief executive officer of the Cystic Fibrosis Foundation in Bethesda, Md. “We look forward to the next stage of clinical development to demonstrate the benefits of this promising therapy.”
Also in the UAB study, PTC124 was shown to be highly selective for fixing only disease-causing mutations, while it spared normal genes, Bedwell said.
The compound has been granted orphan-drug status by the U.S. Food and Drug Administration for the treatment of Duchenne muscular dystrophy and CF, according to the biopharmaceutical company PTC Therapeutics, Inc. of South Plainfield, N.J. It works in an oral form.
Source: University of Alabama at Birmingham