Gene subnetworks predict cancer spread
The metastasis or spread of breast cancer to other tissues in the body can be predicted more accurately by examining subnetworks of gene expression patterns in a patient's tumor, than by conventional gene expression microarrays, according to a presentation at the American Society for Cell Biology (ASCB) 48th Annual Meeting, Dec. 13-17, 2008 in San Francisco.
The subnetworks provide new prognostic markers representing sets of co-functional genes, say scientists at the University of California at San Diego (UCSD), Trey Ideker and Han-Yu Chuang, who worked with Eunjung Lee and Doehaon Lee of the Korean Advanced Institute of Science and Technology in Daejeon, South Korea.
The U.S.-Korea researchers identified the subnetworks by using bioinformatic algorithms to crunch through mountains of gene expression profiles from large cohorts of women with breast cancer.
The data represented women with breast cancer metastasis as well as patients whose tumors had not spread.
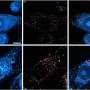
The gene expression profiles were then mapped to the extensive networks of signaling pathways and protein complexes in human cells that had been revealed in previous studies.
Searching the data, the researchers identified subnetworks in which aggregate gene expression patterns distinguished one patient group from another.
They also uncovered many genes associated with breast cancer that had not been identified by previous gene microarray profiles.
Thanks to rapid microarray technology, cancers can now be classified according to their gene expression, or activity patterns.
However, disease classification by gene expression is imprecise because cells taken from a single tumor sample often are heterogeneous; genes switched on in cells from one part of the tumor may not be active elsewhere in the tumor.
In addition, the expression profiles from a range of patients with the "same" type and grade of tumor can differ significantly.
Ideker and Chuang's approach may change diagnostics so that a patient's diagnosis could go beyond, for example, estrogen responsive breast cancer to a particular subtype of estrogen responsive breast cancer with poor or good prognosis.
The U.S.-Korean researchers are now extending their new integrated analysis to other cancers including leukemia, prostate cancer and lung cancer.
They are identifying "condition-responsive" genes within signaling and transcriptional pathways that could be used as a measure of activation levels and could provide another useful tool for diagnosis and prognosis, they say.
Source: American Society for Cell Biology