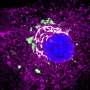
Revealing cancers' weak spots: Researchers exploit genetic 'co-dependence' to kill treatment-resistant tumor cells

(PhysOrg.com) -- Cancer cells fueled by the mutant KRAS oncogene, which makes them notoriously difficult to treat, can be killed by blocking a more vulnerable genetic partner of KRAS, report scientists at the Dana-Farber Cancer Institute and the Broad Institute of Harvard and MIT.
The laboratory results, published today online by the journal Nature in advance of print publication, demonstrate a potential advance against many major tumors that because they harbor the mutant KRAS cancer gene, are highly aggressive and respond poorly to treatment. By targeting the second, more easily inhibited “co-dependent” gene, TBK, the strategy bypasses the so far unfruitful head-on assault against the highly resistant KRAS gene.
“These results represent a new way of targeting oncogenes that have been refractory to standard treatments,” said senior author of the report William Hahn of Dana-Farber, the Broad Institute, and Harvard Medical School (HMS). “What’s particularly exciting is that this approach is potentially highly specific to cancer cells, and therefore should have little toxicity to normal tissues.”
The first author is David Barbie of the two institutes and HMS.
The mutant KRAS oncogene acts like a broken switch, allowing runaway cell growth in nearly all pancreatic tumors, about 25 percent of colorectal, and 25 to 30 percent of lung cancers. When physicians detect KRAS mutations in a cancer, it usually predicts the patient won’t respond well to standard therapies.
“If you have a mutant KRAS, we can’t use many of our newest drugs,” noted Hahn.
For many years, researchers were hopeful that drugs could be designed to shut down KRAS, but this has proven virtually impossible. However, its co-dependent partner, TBK1, encodes a protein kinase — a type of molecular switch for which many inhibitors already exist. TBK1 is not a cancer-causing gene, but in KRAS-driven tumors, TBK1 activity enables cancer cells to survive that otherwise would be destroyed by the body because they are abnormal and dangerous.
TBK1 is the second such gene co-dependent with KRAS to be discovered: In May, a team that included Barbie, Hahn, and other researchers reported in Cell that a kinase gene, STK33, had a similar function in KRAS tumors. Senior author of that report was D. Gary Gilliland then of Brigham and Women’s Hospital, Dana-Farber, the Broad Institute, and HMS. The new approach exploits a relationship between the KRAS and TBK1 genes known as “synthetic lethality.” The term refers to a partnership in which two genes (usually mutated) in a cell have a combined effect that neither has by itself. In some instances, neither mutation alone will kill a cell but the presence of both is lethal.
In the case of KRAS tumors, the opposite is true: both KRAS and TBK1 must be active for the cancer cell to survive; suppressing one or the other kills the cell.
The key role of TBK1 was identified in a large-scale search using the combined resources of Dana-Farber and the Broad Institute to hunt for genes that were essential exclusively to cells with mutant KRAS, but not to cells with wild-type (nonmutant) KRAS or other normal cells.
Working with scientists in the Broad’s RNAi Platform, the team used RNA interference (RNAi) methods to turn off thousands of different genes in 20 laboratory cancer and noncancer cells. They then sought out genes that, when shut down by short pieces of RNA strands, caused KRAS cells to self-destruct, but had no effect on normal cells. The screening process first identified 45 potential candidates, which were further winnowed in a secondary screen to single out TBK1.
“Until four or five years ago, you couldn’t have contemplated doing an experiment like this on so large a scale,” noted Hahn. “We now have the tools that make this possible.” The new paper and its predecessor “really make it clear that you can do this in human cells.”
The discoveries of TBK1 and STK33 are only the first of what the scientists expect will be many more “co-dependent” genes in cancer cells that may prove valuable as drug targets.
“We plan to screen 300 cell lines over the next couple of years,” said Hahn. “The aim is to create a dataset in which any investigator can say, ‘I’m looking for genes that interact with this oncogene or that tumor-suppressor gene. Our intent is to make these data public in order to help move the field forward.”
In the meantime, Hahn said there is considerable interest in testing existing kinase inhibitors as potential new therapies for KRAS tumors. He added that researchers at Dana-Farber and the Broad Institute are interested in developing novel molecular tools to suppress the action of co-dependent oncogene partners.
Provided by Harvard University (news : web)