Breaking oncogene's hold on cancer cell provides new treatment direction
Just as people's bodies and minds can become addicted to substances such as drugs, caffeine, alcohol, their cancers can become addicted to certain genes that insure their continued growth and dominance.
Researchers at Baylor College of Medicine and Harvard Medical School have developed ways to exploit the addictions of cancers to kill them without harming normal tissues. A report on their work appears online today in the journal Science.
Many cancers are driven by the overexpression of oncogenes. These oncogenes are two-faced. On one hand, they promote processes that allow the cells to become immortal and to grow unchecked. On the other hand, the expression of these oncogenes creates additional anti-growth cellular stresses, conflicts that the cancer cell must subvert in order to survive. One classical example of an oncogene that creates such a delicate balance is c-myc. In patients, hyper-activation of c-myc is associated with the most aggressive cancer types, with 20-40 percent of all cancers having an activated myc gene.
"For 30 years, scientists have tried to attack the oncogene myc," said Dr. Thomas Westbrook, assistant professor of molecular and human genetics and biochemistry and molecular biology at BCM and a senior author of the report. "However, it has not been amenable to the drugs we have. Now we have to take advantage of the stresses the oncogene puts on the cancer cell and determine if we can ramp those up to kill the tumor."
"Tumor cells experience considerable mitotic stress," said Westbrook. Regular chemotherapy take advantage of this, but the drugs kill dividing cancer and normal cells. Experts think that special programs within the cancer cell allow it to cope with the stress as it grows and divides (mitosis).
"The fundamental question we asked was how are the stresses in cancer cells different from those in normal cells?" said Westbrook. "We want to exploit that idea and see if we can exacerbate that stress.
To identify genes involved in coping with this stress, Westbrook and his colleague Dr. Stephen Elledge of Harvard Medical School used a special RNA interference screen to disrupt the function of each gene in the genome and identify the genes required to allow the cancer cell to tolerate the stress of the myc oncogene.
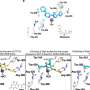
One of the core biochemical processes they uncovered was SUMOylation, a three-step process. Westbrook, Elledge, and colleagues showed that SUMO-activating enzyme, the first step in the process, is required for myc-driven cancers to go through cell division. Thus, inhibiting SAE could be a therapeutic strategy for myc-cancers.
To test this, they turned off the SAE enzyme in a form of myc-driven breast cancer.
"The tumors stopped growing and many of them melted away," said Westbrook. Today, many of the mice are still alive and healthy. If they did not turn off production of the enzyme, the tumors grew and eventually killed the animals.
"If you inhibit this enzyme in a non-myc driven breast cancer, nothing happens," said Westbrook. "If you inhibit it in normal cells of many kinds, nothing happens."
That means that turning off SAE2 exacerbates the stress on cancer cells but not normal cells and thus be a great way to kill cancers without many of the side effects of traditional chemotherapies.
The findings in this report have particular importance for an aggressive form of breast cancer called triple negative breast cancer (TNBC). This subtype is often driven by myc, and there are currently no effective treatments for these patients.
"This may provide that target," said Westbrook. The therapeutic value is that a drug targeting SAE will cause the cancer cell to no longer tolerate myc but will not be detrimental to normal cells.
In addition, myc drives many others kinds of cancers and he anticipates that inhibiting this enzyme in these tumors may have the same effect.
More information: "A Sumoylation-Dependent Transcriptional Subprogram is Required for Myc-Driven Tumorigenesis," Kessler et al. Science, December 8, 2011, vol 334, issue 6061.