Gene identified as a new target for treatment of aggressive childhood eye tumor
New findings from the St. Jude Children's Research Hospital – Washington University Pediatric Cancer Genome Project (PCGP) have helped identify the mechanism that makes the childhood eye tumor retinoblastoma so aggressive. The discovery explains why the tumor develops so rapidly while other cancers can take years or even decades to form.
The finding also led investigators to a new treatment target and possible therapy for the rare childhood tumor of the retina, the light-sensing tissue at the back of the eye. The study appears in the January 11 advance online edition of the scientific journal Nature.
Researchers have known for decades that loss of a tumor suppressor gene named RB1 launches retinoblastoma during fetal development. But the other steps involved in the rapid transformation from a normal cell to a malignant tumor cell that occurs in this cancer were unknown.
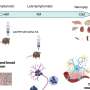
This study linked the RB1 mutation to abnormal activity of other genes linked to cancer without changing the makeup of the genes themselves. Evidence suggests that epigenetic factors, including reversible chemical changes that influence how genes are switched on and off in tumor cells, are altered when RB1 is mutated.
"The dogma in the field has been that once RB1 is mutated, the genome of the affected cell becomes unstable, chromosomes begin to break and recombine, and mutations quickly develop in the pathways that are essential for cancer progression," said Michael Dyer, Ph.D., member of the St. Jude Department of Developmental Neurobiology and a Howard Hughes Medical Institute Early Career Scientist. "What we found through the Pediatric Cancer Genome Project was exactly the opposite. These tumors contain very few mutations or chromosomal rearrangements."
Dyer is one of the paper's corresponding authors. The others are James Downing, M.D., St. Jude scientific director, and Richard Wilson, Ph.D. director of The Genome Institute at Washington University in St. Louis.
Worldwide, retinoblastoma is found in more than 5,000 children each year, including about 300 in the U.S. Most are age 5 or younger, and some are infants when the cancer is discovered, making them among the youngest cancer patients.
While 95 percent of patients are cured with current therapies if their tumors are discovered before they spread beyond the eye, Dyer said the prognosis is much worse for children in developing countries whose cancer is often advanced when it is discovered. For up to half of those patients, retinoblastoma remains a death sentence. Researchers are working to develop curative treatments that preserve vision without radiation or surgical removal of the eye. Success is particularly important for children with tumors in both eyes.
For this study, researchers sequenced the complete normal and cancer genomes of four St. Jude patients with retinoblastoma. The human genome is the complete set of instructions needed to assemble and sustain an individual.
The effort, a first for retinoblastoma, was part of the PCGP that St. Jude and Washington University officials launched in 2010. The three-year project aims to complete whole-genome sequencing of normal and tumor DNA from 600 children and adolescents battling some of the most challenging cancers. Organizers believe the results will provide the foundation for the next generation of clinical care.
The retinoblastoma tumors sequenced contained about 15-fold fewer mutations than have been found in nearly all other cancers sequenced so far. In one patient's tumor, RB1 was the only mutation.
The findings prompted Dyer to integrate the whole-genome sequencing results with additional tests that looked at differences in the patterns of gene activity in tumor and normal tissue. In particular, researchers focused on genes that, when mutated, promote cancer development. "To our surprise and excitement, what we found was that instead of cancer genes having genetic mutations, they were being epigenetically regulated differently than normal cells," Dyer said.
The genes included SYK, which is required for normal blood development and has been linked to other cancers. Drugs targeting the SYK protein are already in clinical trials for adults with leukemia and rheumatoid arthritis.
SYK has no role in normal eye development. When researchers checked SYK protein levels in normal and retinoblastoma tissue, they found high levels of the protein in 82 tumor samples and absent in normal tissue. "We see changes in the SYK gene in retinoblastoma that probably give the cancer cell a growth advantage or provide other key factors with regard to how retinoblastoma is initiated," Wilson said.
When researchers used the experimental drugs to block SYK in human retinoblastoma cells growing in the laboratory or in the eye of a mouse, the cells died. Dyer said work is now underway to reformulate one of the experimental drugs, a SYK-inhibitor called R406, so it can be delivered directly into the eye. If successful, those efforts are expected to lead to a Phase I trial in retinoblastoma patients.
Results of another PCGP study are being published in Nature's Jan. 12 edition. The study provides the first details of the genetic alterations fueling a subtype of acute lymphoblastic leukemia (ALL) that has a poor prognosis. Data from both studies are available at no cost to investigators onthe PCGP Explore website, which can be accessed at http://explore.pediatriccancergenomeproject.org.