Scientists illuminate cancer cells' survival strategy
A team led by scientists at The Scripps Research Institute has discovered key elements of a strategy commonly used by tumor cells to survive when they spread to distant organs. The finding could lead to drugs that could inhibit this metastasis in patients with tumors.
A cell that breaks away from the primary tumor and finds itself in the alien environment of the bloodstream or a new organ, normally is destroyed by a process known as apoptosis. But tumor cells that express high levels of a certain surface protein are protected from apoptosis, greatly enhancing their ability to colonize distant organs. How this protein blocks apoptosis and promotes metastasis has been a mystery—until now.
"What we found in this study is that it's not the increased expression of the protein per se that protects a tumor cell, but, rather, the cleavage of this protein by proteolytic enzymes," said Scripps Research Professor James P. Quigley. "This cleavage triggers a signaling cascade in the tumor cell that blocks apoptosis." Quigley is the principal investigator for the study, which was recently published online before print by the journal Oncogene.
"We think that a reasonable strategy for inhibiting metastasis would be to try to prevent the cleavage of this surface protein using antibodies or small-molecule drugs that bind to the cleavage site of the protein," said Elena I. Deryugina, a staff scientist in Quigley's laboratory and corresponding author of the manuscript.
A Protein Linked to Poor Outcomes
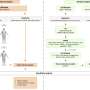
The cell-surface protein at the center of this research is known as CUB Domain Containing Protein 1 (CDCP1). In 2003, a postdoctoral fellow in Quigley's laboratory, John D. Hooper, discovered and co-named CDCP1 as a "Subtractive Immunization Metastasis Antigen," also finding that it is highly expressed on the surfaces of metastasis-prone human tumor cells.
Quigley's laboratory and others soon found additional evidence that CDCP1 plays a major role in enabling metastasis. Clinical studies reported CDCP1 on multiple tumor types and linked its presence to worse outcomes for patients. Deryugina and Quigley reported in 2009 that CDCP1, when expressed in tumor-like cells, strongly promotes their ability to colonize new tissues and that unique monoclonal antibodies to CDCP1, generated in Quigley's lab, significantly block CDCP1-induced tumor colonization. Hooper, who now leads a laboratory at the Mater Medical Research Institute in Brisbane, Australia, reported in a cell culture study in 2010 that most of the CDCP1 protein on the cell membrane could be cleaved by serine proteases. This cleavage event seems to lead to the biochemical activation of the internal fragment of CDCP1 by a process called tyrosine phosphorylation, in this case involving the cancer-linked protein Src.
"What was missing was evidence in live animals that connected CDCP1 biochemically to the blocking of apoptosis and successful metastasis," said Deryugina.
In the new study, Deryugina and her colleagues in the Quigley laboratory, including first author Berta Casar, a postdoctoral fellow, set out to find such evidence.
In Pursuit of Evidence
Hooper supplied the Scripps Research scientists with transformed human embryonic kidney (HEK) cells, which don't naturally express CDCP1, but were forced to express the gene for CDCP1. Casar and Deryugina injected these CDCP1-expressing HEK cells into chick embryos, and found that the CDCP1 proteins on these HEK cells began to be cleaved by resident enzymes to the shorter form. After 96 hours, the proteins were no longer detectable in their full-size, pre-cleaved form. The CDCP1-expressing HEK cells were four times as likely to survive in the chick embryos than were control CDCP1-negative HEK cells. The same results were obtained with HEK cells that express a mutant, non-cleavable form of the CDCP1 protein.
The Scripps Research team then did experiments in live animals with human prostate cancer cells naturally expressing CDCP1 to show that the cleavage of CDCP1 by a serine protease enzyme is the key event that promotes tumor cell survival. "When we blocked CDCP1 cleavage using our unique anti-CDCP1 antibodies, or added a compound that selectively inhibits serine protease enzymes, CDCP1 was not cleaved, and the CDCP1-expressing cancer cells lost almost all their ability to colonize the tissues of chick embryos," said Casar.
Casar and Deryugina also confirmed that in live animals CDCP1's cleavage leads to the biochemical activation of its internal fragment by tyrosine phosphorylation involving the cancer-linked proteins Src and PKCδ. This was followed by the downstream activation of the anti-apoptosis protein Akt and the inhibition of apoptosis-mediating enzymes. The team verified these results with a variety of experimental setups, including tests of tumor-cell lung colonization in mice and tests in which Src signaling was blocked with the anti-Src drug Dasatinib.
Another key experiment by Scripps Research scientists indicated that plasmin, a blood-clot-thinning serine protease, is the principal cleaver of CDCP1 in metastasizing tumor cells. In mice that lack plasmin's precursor molecule, plasminogen, CDCP1-bearing tumor cells showed an absence of CDCP1 cleavage and lost nearly all their ability to survive in lung tissue.
Toward a Promising Strategy
Breakaway tumor cells commonly travel to distant organs via the bloodstream, so their use of an abundant bloodstream enzyme such as plasmin as a survival booster makes sense. "Plasmin has long been linked to cancer," Quigley said. "Unfortunately, it has such an important function in thinning blood clots that using plasmin-inhibiting drugs in cancer patients might do more harm than good."
"Blocking the cleavage of CDCP1 using antibodies or other CDCP1-binding molecules seems to be a more promising strategy," said Deryugina. She and Casar are investigating.
More information: "Blocking of CDCP1 cleavage in vivo prevents Akt-dependent survival and inhibits metastatic colonization via PARP1-mediated apoptosis of cancer cells," www.nature.com/onc/journal/vao … abs/onc2011555a.html