FDA outlines path for lower-priced biotech drugs
(AP) -- The Food and Drug Administration is preparing to review the first lower-cost versions of biotech drugs, expensive medications which have never before faced generic competition.
The guidelines issued by the FDA on Thursday are the final step in a decades-long effort to lower the price of biotech drugs, high-tech injectable medications that cost the nation billions of dollars each year.
"These draft documents are designed to help industry develop biosimilar versions of currently approved biological products, which can enhance competition and may lead to better patient access and lower cost to consumers," FDA's drug division director Dr. Janet Woodcock said in a statement.
Since their introduction in the 1980s, biotech drugs have never faced generic competition because the FDA did not have power to approve copies of such medications. For years the biotech industry successfully argued that their drugs, often made from living cells, were too complex to be duplicated by competitors.
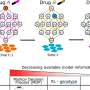
That finally changed with the Obama administration's 2010 health overhaul, which ordered the FDA to create a system for approving so-called "biosimilar drugs." The industry term arose because biotech scientists insisted it would be impossible to produce exact copies of their biologically engineered drugs. They differ from traditional drugs, which are made by combining various chemicals.
Health care data firm IMS Health estimates the global market for biosimilars will range anywhere from $11 billion to $25 billion by 2020, accounting for 4 to 10 percent of the total market for biotech drugs. The Congressional Budget Office estimated biosimilars would save the government $25 billion in reduced health care spending in the coming decade.
The FDA's draft guidelines, posted online, reflect a final agreement that was largely shaped by the biotech industry's demands. As a result, new biotech drugs will enjoy a 12-year period of exclusive, competition-free marketing before a rival medication can launch. Additionally, companies seeking to market biosimilars will have to submit extensive chemical and biological testing data to show that their products function similarly to the originals. The FDA will also have the option to require human and animal clinical studies, the most expensive forms of testing, though staffers said the requirement would be used only when necessary.
"We do not want companies repeating studies that do not need to be done - that wastes precious resources, and of course, exposing humans and animals to unnecessary testing is unethical" said Dr. Rachel Sherman of FDA's office of medical policy. Sherman said the FDA has not yet received any applications for biosimilars drugs, though companies have submitted three dozen requests for meetings on potential products.
The agency will require human testing to declare a biosimilar "interchangeable," with the original drug, a key point of contention between branded drugmakers and their would-be competitors. Such a designation would allow health care professionals to switch patients to a generic version, significantly curbing sales of the original products.
Examples of biotech drugs include specialty cancer drugs like Roche's Avastin, which costs more than $100,000 for a year's supply, and the diabetes staple insulin, which costs closer to $1,000 per year.
Analysts disagree over how much of a discount biosimilars will provide in the U.S. In Europe, where biosimilars have been on the market for several years, knock-off versions of biotech drugs are generally about 20 to 30 percent cheaper than the original products. Companies marketing biosimilars in Europe include Novartis division Sandoz International, Hospira Inc. and Teva Pharmaceutical Industries Ltd. Several U.S.-based brand-name drugmakers have also announced plans to develop biosimilars, including Merck & Co. Inc. and Pfizer Inc.
The FDA said it would take comments on its guidelines for 60 days before finalizing them.
©2012 The Associated Press. All rights reserved. This material may not be published, broadcast, rewritten or redistributed.