'ROCK' off: Study establishes molecular link between genetic defect and heart malformation
. Credit: Joan M. Taylor and Frank L. Conlon, UNC-Chapel Hill.")
UNC researchers have discovered how the genetic defect underlying one of the most common congenital heart diseases keeps the critical organ from developing properly. According to the new research, mutations in a gene called SHP-2 distort the shape of cardiac muscle cells so they are unable to form a fully functioning heart.
The study also shows that treatment with a drug that regulates cell shape rescues the cardiac defect, pointing to therapeutic avenues that could one day benefit Noonan syndrome patients. The results, which were produced in a frog model of the disease, appeared online January 25, 2012, in the journal Development.
Genetic studies have shown that SHP-2 plays a critical role in human physiology and disease. Interestingly, different mutations in different portions of SHP-2 result in three different diseases – Noonan syndrome, a severe congenital heart disease; juvenile myelo-monocytic leukemia, a lethal form of cancer; and Leopard syndrome, a rare condition with skin, facial and cardiac abnormalities. This observation has intrigued a number of researchers, including senior study author Frank Conlon, PhD.
"I've wondered how it is that one mutation gives heart disease and doesn't affect your white blood cells, and another will wipe out your white blood cells and leave your heart alone," said Conlon, an associate professor of genetics and a member of the UNC McAllister Heart Institute. He and others have explored this mystery by creating transgenic animals -- fruit flies, mice, or in Conlon's case, frogs -- that possess a mutated form of SHP-2.
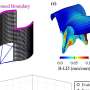
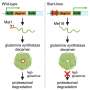
When Conlon and his team genetically engineered frogs to contain the very same defects seen in humans with Noonan syndrome, they found that the frogs did in fact develop cardiac defects. But when they created them with a mutation seen in humans with leukemia, there were no heart defects. The researchers then performed 3D modeling on the animals to assess the nature of the anatomical defects, and discovered that actin filaments – proteins responsible for giving structure to the cardiac muscle cells -- were the ones affected.
Conlon and his collaborator Joan Taylor, PhD, an associate professor of pathology and laboratory medicine at UNC, then tested whether they could reverse the heart malformation using a drug called fausidil that had been shown to improve cardiac function in animal models of heart failure. The drug blocks a protein called ROCK that resides in the same neighborhood – or pathway – of intracellular processes as SHP-2.
The researchers dissolved the drug in the mutant frogs' water tank and found that it did correct the cardiac defects. Their findings connect the dots between Noonan syndrome's underlying genetic defect and the resulting cardiac malformations.
"The human mutations could have been linked to anything, proliferation or cell death, and what this study does is it links it to cell shape changes, which are mediated by this important molecule ROCK," said Conlon. "Our lab studies heart development and heart disease, so we are interested in how this one set of mutations specifically target that one organ. Why the heart? We still have to figure that out."