Fasudil bypasses genetic cause of spinal birth defect
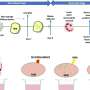
 mice treated with fasudil have larger muscle fibers (right) than SMA mice without fasudil (left). Credit: Melissa Bowerman, Ottawa Hospital Research Institute, University of Ottawa")
Scientists from the Ottawa Hospital Research Institute (OHRI) and the University of Ottawa (uOttawa) have discovered that a drug called fasudil can extend the average lifespan of mice with Spinal muscular atrophy (SMA) from 30.5 days to more than 300 days. The study is published today in BioMed Central's open access journal BMC Medicine, by Dr. Rashmi Kothary, his graduate student Melissa Bowerman and others.
SMA is the leading inherited cause of death in infants and toddlers, affecting approximately 25,000 people in Canada and the United States. Scientists have known for many years that this disease is caused by inherited mutations in a gene called survival motor neuron 1 (SMN1). Most early attempts at developing treatments for SMA focused on replacing this gene, however, Dr. Kothary's group has focused on understanding and targeting the physiological defects present in certain nerve cells with SMA. These cells have a weakened internal scaffold, which hinders their ability to connect with muscle cells and contributes to the severe muscle weakness associated with SMA.
Two years ago, Dr. Kothary and his team showed that a laboratory compound called Y-27632, which targets an enzyme that is involved in maintaining the cellular scaffold, could greatly increase lifespan in a certain mouse model of SMA. In this new study, they tested a compound called fasudil, which is similar to Y-27632, but has the advantage that it has already been approved for human clinical trials for other conditions, meaning that it could possibly be re-targeted to use in clinical trials for SMA more quickly than a completely new drug.
The Kothary group found that fasudil-treated SMA mice survived for an average of more than 300 days, compared to just 30.5 days for untreated SMA mice. However, the average lifespan of fasudil-treated SMA mice was still only about half as long as that of normal mice. Fasudil-treated SMA mice also had larger muscle fibres than the untreated SMA mice, and they behaved more normally with respect to grooming and other regular activities. However, they did not perform any better in strength and balance tests and they still had low numbers of motor neurons, which is typical for SMA.
"Our study is important because it expands a new area of research into SMA, which could lead to the development of new treatments," said Melissa Bowerman. "Of course, this research is still at the early stages and although we found that fasudil could significantly increase lifespan in a mouse model of SMA, this drug couldn't correct all the problems in these mice, and it had serious side effects when used at higher doses."
"A number of groups are working to develop fasudil-like compounds with fewer side effects, and we're very excited to see how these perform in our models, and hopefully in human SMA clinical trials some day," said Dr. Kothary "However, we continue to believe that SMA is a disease that will best be addressed using multiple strategies together, including nutrition and possibly drug, cell and gene therapies."
"Dr. Kothary's group has been a pioneer in SMA research, both in characterizing the impact of SMA on tissues and organs, and in discovering a novel therapeutic pathway involving enzymes that target the cell scaffold," said Dr. Alex MacKenzie, an expert in SMA at CHEO Research Institute and the University of Ottawa, who was not involved in the study. "It has to be said that this approach was not intuitively obvious and Dr. Kothary and his team are to be commended for their creativity in its discovery. It represents an important addition to the armamentarium of experimental SMA treatments."
Although fasudil has been approved by the U.S. Food and Drug Administration for use in certain adult human clinical trials, it is still considered experimental, and has not been approved for the treatment of any human condition in the United States or Canada. Individuals who are interested in experimental therapies should discuss this with their health care professional.
Dr. Kothary is a Senior Scientist at OHRI and a Professor in uOttawa's Faculty of Medicine. He also holds the University Health Research Chair in Neuromuscular Disorders. Melissa Bowerman is a PhD student in Dr. Kothary's group and is a recipient of a Frederick Banting and Charles Best Doctoral Research Award from the Canadian Institutes of Health Research. This research was supported by the Canadian Institutes of Health Research and the U.S. Muscular Dystrophy Association. In addition, all research at OHRI is supported by The Ottawa Hospital Foundation.
More information: Fasudil improves survival and promotes skeletal muscle development in a mouse model of spinal muscular atrophy, Melissa Bowerman, Lyndsay M Murray, Justin G Boyer, Carrie L Anderson and Rashmi Kothary, BMC Medicine (in press)