New treatment shows promise for kids with life-threatening bone disorder
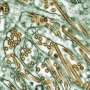
. After 24 weeks of treatment, substantial bone formation is visible in the same hand (right). Credit: The New England Journal of Medicine, copyright 2012")
Doctors at Washington University School of Medicine in St. Louis, working with Shriners Hospital for Children and other institutions, have identified a promising new treatment for a rare and sometimes life-threatening bone disorder that can affect infants and young children.
Known as hypophosphatasia, the condition upsets bone metabolism, blocking important minerals such as calcium from depositing in the skeleton.
In the March 8, 2012, issue of the New England Journal of Medicine, researchers report that at one year of treatment with a new compound, young patients with the most severe forms of hypophosphatasia typically showed greatly improved symptoms, including increased bone strength, better breathing and improved motor development. Some children made enough progress to walk.
"This was a small trial, but we were thrilled to see these results," says first author Michael P. Whyte, MD, professor of medicine, of pediatrics and of genetics at Washington University School of Medicine in St. Louis, who treats patients at Shriners Hospital for Children in St. Louis. "From our experience with studies in mice, we had high hopes. But I think the outcome thus far is beyond anything we had expected."
Hypophosphatasia varies greatly in severity. Its mildest forms may not become apparent until adulthood, and sometimes it may only affect teeth. But in childhood and especially infancy, it can lead to bone weakness, known as rickets. In its most severe forms, hypophosphatasia can lead to death by respiratory failure and has been estimated to occur in about one in 100,000 births. But this number varies worldwide. It is most common in the Mennonite community in Manitoba, Canada, where one in every 2,500 newborns shows severe disease.
"When the condition is extremely severe, a baby may be born with almost no visible bones in an X-ray," says Whyte, also the medical and scientific director of the Center for Metabolic Bone Disease and Molecular Research at Shriners Hospitals for Children in St. Louis. "If an infant has fractured or very thin ribs, the thorax is not going to work properly as a bellows, and respiration is compromised. Together with the profound muscle weakness also seen in severe hypophosphatasia, respiratory lethality is a frequent consequence."
Eleven infants and young children diagnosed with severe hypophosphatasia were recruited to participate in the study. At the beginning, they ranged in age from 20 days to three years. Since there is no approved medical therapy for hypophosphatasia, all 11 patients could receive the experimental treatment, a compound called ENB-0040, also known as asfotase alfa.
At the beginning of the study, nine of the 11 patients had severe or extremely severe rickets; two were classified as moderate. Most patients required respiratory support to help them breathe. Delayed in motor development, most could only turn their heads while lying on their backs. Two of the older children (between 2 and 3 years old) could sit unsupported, but could not crawl or pull to standing.
Nine of the patients completed one year of treatment. One patient withdrew from the treatment, and one patient died due to sudden fever and sepsis that was not attributable to the experimental therapy.
After six months of treatment, most patients showed substantial healing of rickets. After one year, six patients were breathing unaided. Of the nine patients who completed one year of treatment, all made progress, sometimes significant, in motor development. One progressed to moving all limbs against gravity, one to sitting unsupported, two could crawl, one pulled to standing, and two started taking steps. Of the two older children who could only sit, both progressed to walking after a year of treatment.
As with many genetic disorders, hypophosphatasia is caused by mutations that impair an important protein. In this case, the protein is an enzyme called alkaline phosphatase.
Impaired alkaline phosphatase can't break down chemicals as it should. One of the chemicals that builds up as a result is known to inhibit mineralization, blocking calcium and phosphate crystals from growing and entering the skeleton to build normal bone.
The experimental treatment used in this study, ENB-0040, is a manufactured form of normal alkaline phosphatase, but enhanced so that it is targeted to bone.
Treating these patients by giving them normal alkaline phosphatase is not a new idea, Whyte says. More than two decades ago, he and his colleagues at Washington University attempted to treat patients with hypophosphatasia by giving them blood plasma with excess alkaline phosphatase.
That unsuccessful study showed that raising alkaline phosphatase levels in the blood was not enough. More recently, Whyte's industry collaborators have provided the missing link: Adding a short protein chain that adheres to bone allowed the alkaline phosphatase to be targeted to the skeleton.
Paving the way for this human study, Whyte and his colleagues then showed that the targeting chain worked well in a mouse model of severe hypophosphatasia, restoring normal life span to mice, as long as they received daily injections of ENB-0040 starting at birth.
The nine patients who completed one year of treatment continue to receive therapy and are now participating in an extension study. For more information about clinical trails recruiting patients with hypophosphatasia, visit clinicaltrials.gov.
More information: Whyte MP, Greenberg CR, Salman NJ, Bober MB, McAlister WH, Wenkert D, Van Sickle BJ, Simmons JH, Edgar TS, Bauer ML, Hamdan MA, Bishop N, Lutz, RE, McGinn M, Craig S, Moore JN, Taylor JW, Cleveland RH, Cranley WR, Lim R, Thacher TD, Mayhew JE, Downs M, Millan JL, Skrinar AM, Crine P, Landy H. Enzyme-replacement therapy in life-threatening hypophosphatasia. New England Journal of Medicine. Vol 366. Pages 904 – 913. March 8, 2012.
Millan JL, Narisawa S, Lemire I, Loisel TP, Boileau G, Leonard P, Gramatikova S, Terkeltaub R, Camacho NP, McKee MD, Crine P, Whyte MP. Enzyme replacement therapy for murine hypophosphatasia. Journal of Bone and Mineral Research. November 2008.