Phylogenomic analysis reveals origin, spread of MRSA clone
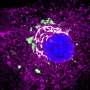
(HealthDay) -- Phylogenomic analysis has revealed details about the emergence and transmission of a major methicillin-resistant Staphylococcus aureus (MRSA) clone, EMRSA-16, according to research published online May 14 in the Proceedings of the National Academy of Sciences.
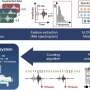
Paul R. McAdam, from the University of Edinburgh in the United Kingdom, and colleagues performed a Bayesian phylogenetic reconstruction on the basis of genome sequences from 87 Staphylococcus aureus isolates. The isolates were collected from patients in three continents over a 53-year period and included 60 isolates of the pandemic EMRSA-16 clone and 27 additional clonal complex 30 (CC30) isolates.
The researchers found that there was a common ancestor shared by the three major pandemic clones, originating from the CC30 lineage (phage type 80/81, Southwest Pacific, and EMRSA-16), which existed more than 100 years ago. In contrast, the hospital-associated EMRSA-16 clone likely emerged 35 years ago. A genome-wide analysis of CC30 revealed molecular correlates of hospital- or community-associated pandemics, including mobile genetic elements and nonsynonymous mutations impacting both antibiotic resistance and virulence. Phylogeographic analysis demonstrated that the spread of EMRSA-16 within the United Kingdom was from hospitals in large population centers in London and Glasgow to regional health care settings.
"Taken together, the high-resolution phylogenomic approach used resulted in a unique understanding of the emergence and transmission of a major MRSA clone and provided molecular correlates of its hospital adaptation," the authors write.
ARK-Genomics at the Roslin Institute performed sequencing services for the study.
More information:
Abstract
Full Text (subscription or payment may be required)
Copyright © 2012 HealthDay. All rights reserved.