Study reveals how the world's first drug for amyloid disease works
")
Scientists from The Scripps Research Institute and Pfizer Inc. have published a new study showing how a new drug called tafamidis (Vyndaqel) works. Tafamidis, approved for use in Europe and currently under review by the US Food and Drug Administration (FDA), is the first medication approved by a major regulatory agency to treat an amyloid disease, a class of conditions that include Alzheimer's.
Tafamidis treats a deadly nerve disease caused by transthyretin (TTR) amyloid fibril formation, or the accumulation of abnormal assemblies of the TTR protein. The drug inhibits TTR aggregation, and clinical trials have shown that it delays the typical progression of nerve destruction in polyneuropathy patients.
"The details in this new paper, combined with clinical trial data, show for the first time that an amyloid disease can be successfully treated by reducing the rate of amyloid formation," said Jeffery W. Kelly, chair of the Department of Molecular and Experimental Medicine, the Lita Annenberg Hazen Professor of Chemistry, and member of the Skaggs Institute for Chemical Biology at Scripps Research. Kelly is a senior author of the new paper, which appears in an advance, online Early Edition issue of Proceedings of the National Academy of Sciences on May 29, 2012.
An Array of Progressive Symptoms
While the naturally occurring or "wild type" transthyretin protein is prone to aggregate in older people causing cardiac disease, a variety of destabilizing mutations lead either to a primary cardiomyopathy or to early onset forms of polyneuropathy, known as TTR familial amyloid polyneuropathy, affecting about 10,000 people worldwide.
Familial amyloid polyneuropathy compromises the peripheral and autonomic nervous systems, with symptoms including sensory deprivation and pain, muscle weakness and wasting, and alternating constipation and diarrhea. In some familial amyloid polyneuropathy patients, cardiomyopathy can present later in the course of the disease.
In transthyretin amyloid diseases that present primarily as a cardiomyopathy, doctors have been able to stave off heart failure with a liver and heart transplant; familial amyloid polyneuropathy patients receiving a liver transplant can benefit, since the liver is the primary source of mutant, disease-associated TTR. For the 90 percent of patients surviving transplantation, this surgical form of gene therapy slows familial amyloid polyneuropathy progression, but does not stop it as the wild type transthyretin protein can continue to form amyloid.
Left untreated, the TTR amyloidoses are relentlessly progressive and inevitably fatal, with a course of about a decade from initial symptoms to death.
The Search for Treatments
Kelly began searching for TTR-amyloidogenesis-inhibitors in the mid 1990s, and a few years later began to focus on a family of TTR-binding compounds, the so-called benzoxazoles, whose basic design would further be elaborated into tafamidis using a structure-based drug design paradigm. In 2003, Kelly co-founded a Cambridge, Massachusetts-based biotechnology startup, FoldRx Pharmaceuticals (now a fully owned subsidiary of Pfizer), to develop these compounds and optimize one of them into an orally available drug for the treatment of the TTR amyloidoses. The result was tafamidis meglumine, whose preclinical tests remained unpublished until now.
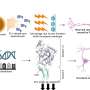
Kelly and his colleagues designed tafamidis to bind to the natural, functional TTR structure (mutant and wild type), in a way that prevents it from deviating from this natural, functional form into the amyloid state. TTR's natural, functional form is a "tetramer" made from four copies of the protein. Amyloidosis occurs when these tetramers come apart and the individual TTR proteins ("monomers") undergo shape changes enabling them to misassemble into dysfunctional amyloid aggregates. Included in the TTR aggregate distribution are amyloid fibrils—protein stacks made from millions of TTR monomers—although researchers suspect that smaller, shorter-lived pre-amyloid aggregates do more direct damage to nerve cells and nerve fibers.
The early onset TTR amyloidoses are caused by inherited TTR mutations that weaken the tetramers' ability to stick together, producing monomers more likely to aggregate into amyloids and other aggregate structures. Fortunately, the TTR tetramer, which is the backup carrier of the thyroid hormone thyroxine through the bloodstream, has two unoccupied thyroxine-binding sites along its longest and weakest seam. Kelly and his colleagues designed tafamidis to grab either of these thyroxine-binding sites, in a way that bridges the seam and helps keep the tetramer from coming apart.
A Stabilizing Influence
The newly published molecular and structural data show that tafamidis does indeed stabilize TTR tetramers, under normal physiological conditions in the bloodstream and even under abnormal conditions when they would be much more likely to fall apart and reassemble as amyloids. Tafamidis has this stabilizing effect on tetramers of the normal "wild-type" TTR protein as well as on those made from disease-associated mutant and wild type TTR subunits.
"There are more than a hundred TTR mutations that cause amyloidosis, but the vast majority of those TTRs are capable of being bound by tafamidis and held in the natural tetramer state," said Kelly.
Throughout the development of tafamidis-type compounds, Kelly and his colleagues collaborated with the Scripps Research laboratory of Ian A. Wilson, who is Hansen Professor of Structural Biology and a member of the Skaggs Institute at Scripps Research. The Wilson laboratory specializes in the use of X-ray crystallography to determine the atomic structures of interacting proteins. Whenever a small molecule stabilizer of TTR was generated that afforded interesting biochemical stabilization, Wilson's team analyzed its structure. "By the end, we had determined more than 30 small molecule stabilizer–TTR structures, in an effort to generate tafamidis and identify the molecular interactions that lead to stabilization of the natural TTR tetramer," said Stephen Connelly, a Wilson laboratory staff scientist who performed these structural studies and who was a co-author the paper.
The small molecule benzoxazole, tafamidis, that ultimately entered clinical trials was optimized for several criteria, including its ability to stabilize the TTR tetramer's weakest seam. "We found that one end of the tafamidis structure fits neatly into the tetramer's hydrophobic thyroxine-binding pocket, while at the other end it binds to nearby polar amino acids, both types of interactions bridging or stabilizing the two halves of the tetramer," Connelly said. The drug's stabilizing force greatly reduces the rate at which these tetramers come apart, and in so doing greatly reduces the rate of amyloid formation.
Tafamidis is considered an "orphan" drug because its initial intended treatment population is the relatively small polyneuropathy population. However, even wild-type TTR forms amyloid in 10 to 20 percent of the growing elderly population—leading to cardiomyopathy. Thus, this condition, and drugs such as tafamidis that can treat it, could be of growing interest to the pharmaceutical industry.
More information: www.pnas.org/content/early/201 … /1121005109.abstract