New genetic mechanism for controlling blood cell development and blood vessel integrity found
The protein GATA2 is known as a "master regulator" of blood cell development. When a mutation occurs in the gene that makes GATA2, serious blood diseases such as acute myeloid leukemia can result.
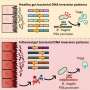
Zooming in on the GATA2 gene, UW-Madison researchers and their collaborators at the National Institutes of Health (NIH) have discovered unexpectedly that a small DNA sequence drives this powerful master regulator.
The sequence plays an essential role in controlling GATA2 production and generating self-renewing blood stem cells responsible for the earliest steps in the development of blood cells of all kinds—red cells to transport oxygen and white cells to fight infection.
The researchers also found that the DNA sequence, which they call the +9.5 GATA2 switch site, ensures that blood vessels function properly to prevent hemorrhaging. Until now, GATA2 had not been implicated in blood vessel integrity. The study appears in The Journal of Clinical Investigation (online Sept. 10, 2012).
Although the study was performed in mice, it should have significant clinical relevance, particularly to physicians and scientists aiming to understand certain types of leukemia and related disorders involving disruptions in the blood and immune systems. The research indicates that downstream genes are impacted when the switch site is altered, leading to abnormal development of the adult blood system, says senior author Dr. Emery Bresnick, professor of cell and regenerative biology at the School of Medicine and Public Health.
"There's every reason to believe that we can use these findings as a foundation to discover key factors and signals that can be modulated therapeutically for the treatment of specific blood and blood vessel disorders," he says.
Bresnick has studied GATA proteins for more than a decade. Among other things, his team discovered five "hot spots" on the GATA2 gene where special activity involving both GATA1 and GATA2 appeared to be taking place. They named these GATA switch sites. Over the last four years, the group has focused on their third GATA2 switch site, +9.5, a small sequence of about 25 base pairs located in a region of the gene called the intron.
"While introns can contain regulatory sequences that control gene activity, until now we and others have been unable to find the mechanisms that control the GATA2 gene, despite years of studying this problem," Bresnick says.
Senior scientist Dr. Kirby Johnson from the Bresnick laboratory led experimental efforts to genetically engineer and breed mice in which the site was knocked out, and then studied the consequences of the mutation.
Without the sequence, the embryos died on day 14, but before that they developed massive hemorrhaging. Closer analysis showed that large veins in the embryo leaked embryonic red blood cells.
"We knew GATA2 was expressed in endothelial cells that line blood vessels, but we didn't know what it was doing there," Bresnick says. "Now we have identified examples in which the endothelial lining of large veins in the embryos was discontinuous."
While it was obvious from the hemorrhaging that the knock-out mice could still produce embryonic red blood cells very early in development, it was not clear if this capability continued later in development to produce the diverse array of adult blood cells. So the UW team turned its attention to stem cells and progenitor cells in the embryonic yolk sac and fetal liver, where blood production, or hematopoeisis, takes place.
Using a robust test for progenitor cells, they found that activity was not compromised in the yolk sac, which provides embryonic red blood cells to the early embryo, but defects were seen later in development in the fetal liver, a major site of blood stem and progenitor cell production.
"We were grateful to be able to collaborate with Dr. Jing Zhang, a stem cell scientist based at UW's McArdle Laboratory for Cancer Research who routinely studies hematopoietic stem cell activity using the 'gold standard' approach of cell transplantation," Bresnick says.
Zhang analyzed the fetal liver compartment and found that stem cells were completely missing.
"These assays informed us that the +9.5 switch site is absolutely required for GATA2 to be expressed in the fetal liver and for the establishment and/or maintenance of hematopoietic stem cells," Bresnick says.
Although the UW team's analyses were performed using mouse models, Bresnick's NIH collaborators identified the same switch site mutation in a patient with MonoMAC syndrome, a rare immunodeficiency disease characterized by blood and immune cell disorders and increased susceptibility to mycobacterial disease, warts and certain cancers. The syndrome typically is caused by a mutation in the gene coding region of GATA2. A number of people with MonoMAC syndrome are enrolled in clinical studies overseen by Dr. Steven Holland, chief of the Laboratory of Clinical Infectious Diseases at the National Institute of Allergy and Infectious Disease, part of the NIH.
After identifying four people with MonoMAC syndrome who lacked the usual mutation, Amy Hsu, an investigator in Holland's laboratory, sequenced the +9.5 GATA2 switch site in the patients. She found that one of them had a deletion in the switch site that caused blood vessel disorders in the mice from Bresnick's laboratory.
"Given the importance of GATA2 for controlling blood cell development, I was not surprised that alterations in any portion of the GATA2 gene would be linked to a blood disorder," Bresnick says.
"But finding that the small +9.5 sequence we discovered was disrupted in the human disease MonoMAC was quite striking."
It may be the only example in which a small DNA disruption in a regulatory region of a gene produces such a profound effect as eliminating adult stem cells, he adds.
"We feel that the +9.5 sequence's ability to produce stem cells, combined with its critical role in conferring blood vessel integrity, has earned it the description of a master regulatory GATA switch site," he says.