Scientists dramatically reduce plaque-forming substances in mice with Alzheimer's disease
Scientists have found that eliminating an enzyme from mice with symptoms of Alzheimer's disease leads to a 90 percent reduction in the compounds responsible for formation of the plaques linked to Alzheimer's disease.
That is the most dramatic reduction in this compound reported to date in published research.
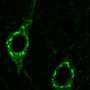
The compounds are amyloid beta, or A-beta peptides; peptides are proteins, but are shorter in length. When A-beta peptides accumulate in excessive amounts in the brain, they can form plaques, which are a hallmark of Alzheimer's disease.
"These mice are models for the most aggressive form of Alzheimer's disease and produce the highest amount of A-beta peptides. This 90 percent reduction is the biggest drop in A-beta levels that has been reported so far by treating animal models with drugs or genetic manipulations," said Sung Ok Yoon, associate professor of molecular & cellular biochemistry at Ohio State University and lead author of the study.
The key to reducing A-beta peptides was the elimination of an enzyme called jnk3. This enzyme stimulates a protein that produces A-beta peptides, suggesting that when jnk3 activities are high, A-beta peptide production increases – increasing chances for their accumulation and formation into plaques.
The researchers also observed that jnk3 activities in brain tissue from Alzheimer's disease patients were increased by 30 to 40 percent when compared to normal human brain tissue. Jnk3 activity typically remains low in the brain, but increases when physiological abnormalities arise.
The findings suggest that jnk3 could be a new target for Alzheimer's disease intervention, Yoon said. So far, some drugs can slow the disease's progression, but there is no cure.
The research is published in the Sept. 6, 2012 issue of the journal Neuron.
Alzheimer's disease affects more than 5 million Americans, and its cause remains unknown. Although scientists have not yet determined whether A-beta peptides present in plaques cause Alzheimer's disease or form as a consequence of the disease, the presence of the plaques is linked to progressive cognitive decline.
In this study, Yoon and colleagues deleted jnk3 genetically from Alzheimer's disease model mice carrying the mutations that are found among early-onset Alzheimer's disease patients. In six months, the deletion of the enzyme had lowered A-beta peptide production by 90 percent, which persisted over time, with a 70 percent reduction seen at 12 months in these mice.
When the researchers saw that elimination of jnk3 dramatically lowered A-beta peptides in the mice, they also looked for effects on cognitive function at 12 months. The deletion of jnk3 improved cognitive function significantly, reaching 80 percent of normal, while cognitive function in disease model mice was 40 percent of normal. The number of brain cells, or neurons, in the Alzheimer's disease mice was also increased with jnk3 deletion, reaching 86 percent of the value in normal mice, while the neuron numbers were only 74 percent in Alzheimer's model mice.
Jnk3 is an enzyme that modifies its target proteins, changing the protein properties. Amyloid precursor protein (APP), which produces A-beta peptides, was already known to be modified in Alzheimer's disease brains. Yoon and colleagues also found that jnk3 modifies APP, which leads to stimulation of A-beta peptide production.
The scientists examined whether patterns of RNA expression in the Alzheimer's disease mouse brains were changed when jnk3 was deleted. This pattern tells scientists whether cells are behaving as expected. The results were a big surprise: Expression of genes that are necessary for new protein production, or synthesis, was significantly reduced in the Alzheimer's model brains compared to normal mouse brains.
"A lot of neurons had shut off their protein production. And when we deleted jnk3, the neurons' overall protein production came very close to normal levels," Yoon said.
Experiments in neuron cultures circled this phenomenon back to high levels of A-beta peptides, showing that A-beta peptides turn off new protein production by activating another enzyme called AMP kinase (AMPK). AMPK is normally activated when cells are starved of nutrients, such as right before a meal. For that reason, AMPK is a popular target in diseases associated with the body's use of glucose and fats for metabolism, such as Type 2 diabetes.
The researchers observed that once activated, AMPK eventually silenced a powerful sequence of chemical reactions called the mTOR pathway, which controls new protein synthesis in a variety of cell types. This phenomenon launches a stress response in the ER (endoplasmic reticulum), which is the protein synthesis machine present in every single cell.
"The interesting thing is, it had already been published that when ER stress is induced, that could activate jnk3," Yoon said.
As a result, she and her colleagues proposed a model to describe their hypothesis. Continual jnk3 activation by ER stress allows a detrimental cycle to start, and this cycle gets stronger over time as follows: An as-yet unidentified physiological problem increases jnk3 activity, which leads to initial production of A-beta peptides from APP. These peptides stimulate the AMPK enzyme. AMPK then blocks new protein production by the mTOR pathway. The lowered protein production leads to ER stress, and this increases jnk3 activities. As at the start, the increased jnk3 activities lead to production of more A-beta, adding "more push" to the cycle, Yoon explained.
"So, around and around and around it goes, ever more strongly. These results suggest that jnk3 is the key perpetuating the cycle," she said.
To test the hypothesis, the researchers treated live mouse brain tissues with one drug that blocks the mTOR pathway or another drug that induces ER stress. Both treatments dramatically increased A-beta peptide production within nine hours, but only when jnk3 was present. And revisiting the human data, the researchers also observed that Alzheimer's disease brain tissue showed a prominent elevation of ER stress.
Though a missing link remains – the pathological condition that produces this stress in the first place – Yoon said the demonstration that A-beta peptides block new protein production reveals new ways of thinking about Alzheimer's disease treatment.
"The fact that we found that protein synthesis is hugely affected by Alzheimer's disease opens up a door to let us try a variety of drugs that are already developed for other chronic progressive diseases that share this commonality of affected protein production," Yoon said.
For example, many cancer drugs are designed to block new protein synthesis to slow protein production among cancer cells. Even though the opposite effect is desired in Alzheimer's disease, these drugs may represent a starting point for a new class of Alzheimer's disease therapies, she said.
Yoon also hopes to test whether small-molecule jnk3 inhibitors could potentially improve cognitive function in Alzheimer's disease mouse models.