Drug reverses abnormal brain function in Rett syndrome mice
A promising study out today in the prestigious Journal of Neuroscience showed that in a mouse model of Rett syndrome, researchers were able to reverse abnormalities in brain activity and improve neurological function by treating the animals with an FDA-approved anesthesia drug, ketamine. Rett syndrome is among the most severe autism-related disorders, affecting about one in 10,000 female births per year, with no effective treatments available.
"These studies provide new evidence that drug treatment can reverse abnormalities in brain function in Rett syndrome mice," says David Katz, PhD, professor of neurosciences, Case Western Reserve University School of Medicine and senior author of the study. "They also provide new leads as to what kinds of drugs might be effective in individuals with Rett syndrome."
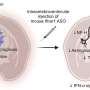
Neuroscientists at Case Western Reserve University School of Medicine were able to successfully map differences in the brain activity of normal mice and those with a genetic mutation that mirrors the cause of Rett syndrome in humans. They found that – compared to normal mice – Rett syndrome mice showed regions of abnormally low activity in the front of the brain (forebrain) and regions of abnormally high activity in the back of the brain (brainstem). Importantly, they found that the regions of low activity overlap with regions of the brain that are also under-active in humans with classic autism. This indicates there may be common mechanisms underlying abnormal behaviors in the two diseases.
The identification of these brain regions provided clues into specific areas to target for treatment. Based on previously published findings that ketamine activated neurons in the forebrain, the researchers gave the drug to the Rett syndrome mice and found it increased levels of brain activity in those regions and improved neurological function. Importantly, the drug was effective at a low dose that did not produce anesthesia.
Katz strongly cautioned that, because ketamine can have potent anesthetic effects and is a controlled substance, further work is needed to establish the safety of ketamine in patients with Rett syndrome. Moreover, ketamine has never been used to treat a chronic condition, and additional studies are required to determine whether or not this is feasible and safe. However, safer drugs acting in the same pathways as ketamine may be available.
Unlike most disorders on the autism spectrum, researchers know the cause of Rett syndrome – a genetic change on the X chromosome, which helps explain why it affects girls almost exclusively. Families don't usually know if a newborn has Rett syndrome because affected children can appear normal for the first six to 18 months after birth. Then, parents start to notice the infant losing the ability to speak, move, eat or even breathe normally. Many girls with Rett syndrome can live into adulthood and are so disabled that they require round-the-clock care.
One in 88 Americans is affected by an autism-related disorder, according to the Centers for Disease Control. Those affected by Rett syndrome can lose – to varying degrees – the ability for normal human interaction. They can be socially withdrawn, struggle to communicate and tend to engage in repetitive behaviors – all hallmarks of disorders that fall within the autism spectrum.
Katz's team in the School of Medicine included post-doctoral fellows Miriam Kron, PhD and Michael Ogier, PhD, research assistants C. James Howell and Ian Adams and undergraduate students Michael Ransbottom and Diana Christian. Kron and Howell are the lead authors on the Journal of Neuroscience paper.
The findings were supported by grants from the National Institute of Neurological Diseases and Stroke at the National Institutes of Health (NS-057398), the International Rett Syndrome Foundation and the first ever grant awarded in Northeast Ohio by Autism Speaks, the world's leading autism science and advocacy organization.