Key discovered to how chemotherapy drug causes heart failure
Doxorubicin, a 50-year-old chemotherapy drug still in widespread use against a variety of cancers, has long been known to destroy heart tissue, as well as tumors, in some patients.
Scientists have identified an unexpected mechanism via the enzyme Top2b that drives the drug's attack on heart muscle, providing a new approach for identifying patients who can safely tolerate doxorubicin and for developing new drugs. A team led by scientists at The University of Texas MD Anderson Cancer Center reports its findings about the general DNA-damaging drug today in the journal Nature Medicine.
"Even in this age of targeted therapies, doxorubicin remains an effective agent used mainly in combination with other drugs against a variety of malignancies, including breast, lung, ovarian and bladder cancers, as well as leukemia and lymphoma," said Edward T.H. Yeh, M.D., professor and chair of MD Anderson's Department of Cardiology and senior author of the study.
"However, its use is limited by its cardiotoxicity, which can lead to heart failure," Yeh said. "We're excited because we've identified the molecular basis for doxorubicin's damage to the heart."
A tale of two enzymes
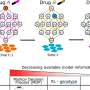
Doxorubicin binds to topoisomerase2 (Top2), an enzyme that controls the unwinding of DNA necessary for cell division.
There are two types of Top2, Yeh said. Top2a is overproduced in cancer cells but largely absent in normal cells. The reverse is true for Top2b, virtually absent in cancer cells but present in normal cells.
Doxorubicin destroys cancer cells by binding to Top2a and to DNA, causing irreparable damage in the form of double-strand DNA breaks. This triggers apoptosis, a cellular suicide mechanism designed to prevent the growth of defective cells.
Yeh and colleagues found that the drug binds to Top2b in cardiomyocytes - heart muscle cells - but it inflicts its damage in a different manner from its attack on cancer cells, yet consistent with longstanding belief about the heart-damaging culprit.
Old suspect: reactive oxygen species
Increases in reactive oxygen species (ROS), highly reactive molecules that contain oxygen, have been observed after doxorubicin treatment. ROS are a normal byproduct of metabolism and play other roles, but at high levels cause cellular damage, a condition called oxidative stress.
ROS damage to cardiomyocytes via the redox cycle - a swapping of electrons to cause either oxidation or reduction of molecules - was hypothesized as the cause of doxorubicin-driven cardiotoxicity. Yet, therapies to directly reduce ROS levels did not prevent heart damage.
"We provide an explanation for the classic observation that doxorubicin generates major ROS, but we show that the entire cardiotoxicity cascade depends on Top2b," Yeh said.
The experiments
The team developed an inducible mouse model in which treatment with the drug tamoxifen would knock out the Top2b gene only in heart muscle. They found:
- Top2b protein levels were much lower in the knockout mice.
- Top2b is not necessary for heart health. Mice without the gene lived for more than 10 months in excellent health.
Then they treated mice with and without Top2b with doxorubicin and analyzed their hearts 16 hours later. The results from microarray analysis include:
- Activation of DNA damage control and apoptosis genes, including the p53 pathway, was greatly increased in treated mice with intact Top2b.
- Increased levels of Top2b correlated with increases in gene transcription, double-strand breaks and cell death.
So far, it looked a lot like the way doxorubicin attacks cancer cells via Top2a. But the team then repeated the experiment 72 hours after doxorubicin treatment. They found:
- Activation of DNA damage pathways was replaced by mitochondrial dysfunction and oxidative phosphorylation pathways in mice with intact Top2b. Mitochondria generate a cell's energy and in the process control ROS.
- Expression of genes vital to the formation and proper function of mitochondria was reduced in the presence of Top2b.
New suspect: Topoisomerase 2b
A series of experiments confirmed that ROS generation was caused by changes in gene activation, not the redox cycle; that doxorubicin treatment generated ROS in the hearts of Top2b-positive mice, but this was reduced by 70 percent in mice with Top2b knocked out; and that hearts of mice with intact Top2b had diminished pumping capacity after treatment with the drug.
Thus, doxorubicin causes heart damage both by inducing DNA double strand breaks and by affecting the heart muscle's metabolism. Both factors are entirely dependent on Top2b.
Clinical study launched to test biomarker potential
The team's mouse model experiments led to a clinical study now under way among two types of cancer patients - those who have received small amounts of doxorubicin and developed heart problems, and those who received large amounts of the drug yet without apparent heart damage.
The study aims to find whether patients' blood levels of Top2b indicate their sensitivity to doxorubicin-induced heart damage. It's funded by a $1.84 million, 5-year grant from the Cancer Prevention and Research Institute of Texas.
If the outcome of the clinical study is as predicted, a simple blood test could indicate who will be sensitive to doxorubicin, Yeh said. Protective measures, such as using cardiac protective drugs or close monitoring, could be taken early in treatment or the drug could be avoided altogether.
Another exciting alternative to avoid doxorubicin-induced cardiotoxicity is to develop drugs that only target Top2a, Yeh said.
"We want to make sure that cancer patients will have healthy hearts to enjoy their life after successful cancer treatment," Yeh said.