TGen professor discusses benefits of whole genome sequencing in study of multiple myeloma
The scientific benefits of whole genome sequencing at the Translational Genomics Research Institute (TGen) will be presented at the 14th International Myeloma Workshop, April 3-7 at the Kyoto International Conference Center.
Dr. Jonathan Keats, head of TGen's Multiple Myeloma Research Laboratory, will present Discovering the Underlying Genetics of Multiple Myeloma Through Whole Genome Sequencing at 8:15 a.m. (Kyoto time) April 4, following the conference opening talk.
Multiple myeloma is a pathological description of a disease characterized by the accumulation of plasma cells in the bone marrow. Dr. Keats' lab at TGen is focused on using new methods to investigate the genomic features of this disease with the goal of identifying genetic events that drive the development, progression, and mediate therapeutic resistance.
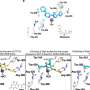
"We will show for the first time the integration of DNA and RNA sequencing in multiple myeloma, and how TGen's comprehensive approach to this research has begun to uncover possible genetic changes that could lead to the underlying causes of this cancer," Dr. Keats said.
Previous studies have identified as many as 10 distinct biological subgroups of multiple myeloma, highlighting the need to identify distinct genetic defects to address each subtype of this disease.
Recent advances in next generation sequencing can now identify nearly all genetics events existing in an individual tumor. Initial studies have focused on whole genome sequencing or exome sequencing and confirmed genetic mutations (TP53, NRAS, KRAS) as well as identified novel mutations (FAM46C and DIS3). In addition, identification of recurrent BRAF mutations and the availability of targeted BRAF inhibitors provide an opportunity to translate research findings into clinical practice to benefit patients.
Dr. Keats is one of the key researchers in TGen's Multiple Myeloma Genomics Initiative, funded by the Multiple Myeloma Research Foundation.
"We will present results from the multiple myeloma genomics initiative using paired whole genome and transcriptome sequencing on 84 patient samples and 68 cell lines. The combination of DNA and RNA based sequencing approaches has improved our ability to identify biologically relevant alterations within each sample," Dr. Keats said.
"Additional analysis will improve out understanding of what leads to multiple myeloma and hopefully lead to new and improved classification and prognostic models."