Scientists find clues to some inherited heart diseases
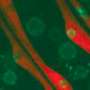
, MKL1 is located in the cell body; upon stimulation, it moves into the cell nucleus (appears white), where it helps to turn on genes required for the cell's adaptation to mechanical stress. In contrast, in cells from a mouse model of muscular dystrophy (bottom), MKL1 remains outside the nucleus, failing to activate the normal gene response. Credit: Chin Yee Ho")
(Medical Xpress)—Cornell researchers have uncovered the basic cell biology that helps explain heart defects found in diseases known as laminopathies, a group of some 15 genetic disorders that include forms of muscular dystrophy and between 5 percent and 10 percent of all cases of inherited heart disease.
The findings, published online May 5 in Nature, reveal possible places in a biological pathway where drug therapies may eventually be applied to treat the heart disease associated with such diverse diseases as Emery-Dreifuss muscular dystrophy, dilated cardiomyopathy and limb-girdle muscular dystrophy.
Many laminopathies, caused by mutations to the so-called LMNA gene, affect skeletal and cardiac muscles for reasons that have not been well understood. The LMNA gene codes for the proteins lamin A and C, which provide stability to the cell's nucleus and play important roles in gene regulation.
In order to study heart failure associated with laminopathies, the researchers made use of existing mouse models – mice bred for research that may have an existing, inbred or induced disease that is similar to a human condition. One of the mouse models has mutations in the LMNA gene that produces mice with enlarged hearts that failed, leading to death at eight to 10 weeks of age. Another mouse model used in the study completely lacks lamins A and C, and the mice develop muscular dystrophy with similar heart conditions that cause death at around four to eight weeks of age, said Jan Lammerding, the paper's senior author, assistant professor in the Weill Institute for Cell and Molecular Biology and the Department of Biomedical Engineering.
In normal cells, a signaling protein called MLK1 resides in the cell's body, outside of the nucleus. MLK1 binds to a protein called G-actin, which covers up a code on MLK1's surface that permits entry into the cell's nucleus. When the cell is mechanically stressed or exposed to specific growth factors, "an adaptive response occurs to reinforce the muscle cell to be fully functional," said Lammerding.
In the process, G-actin detaches from MKL1 and is converted into another protein called F-actin, which exposes the code for MLK1's entry into the nucleus. Once inside the nucleus, MLK1 binds to another protein to activate genes for reinforcing the cell's structure.
In the experimental mice, G-actin failed to consistently convert to F-actin. As a result, G-actin often remained bound to MKL1, blocking its entry code. The culprit behind this disruption was a protein called emerin, which triggers processes that sequester MLK1 in the nucleus and is normally held in place at the nucleus' inner membrane by lamins A and C. When the lamins were missing or mutated, emerin floated away from the inner nuclear membrane, and MLK1 was quickly exported out of the nucleus. Without MLK1 in the nucleus, the genes that trigger the cell's adaptations to mechanical stress, including the conversion of G-actin to F-actin, remain inactive, leading to the heart disease found in many laminopathies.
With this knowledge, researchers may eventually create "drugs that modulate and target downstream consequences by stimulating the impaired pathway and turning it on," Lammerding added.
More information:
Ho, C. et al. Lamin A/C and emerin regulate MKL1–SRF activity by modulating actin dynamics, Nature (2013)
doi:10.1038/nature12105