Cell maturity pathway is deleted or weak in glioblastoma multiforme
A program that pushes immature cells to grow up and fulfill their destiny as useful, dedicated cells is short-circuited in the most common and deadly form of brain tumor, scientists at The University of Texas MD Anderson Cancer Center report this week in the Early Edition of the Proceedings of the National Academy of Sciences (PNAS).
Stuck in what amounts to cellular adolescence, these precursor cells accumulate, contributing to the variability among glioblastoma multiforme (GBM) cells that make it so difficult to treat, said first author Jian Hu, Ph.D., instructor of Genomic Medicine.
"This arrested development is driven by the GBM cells' plasticity—their stem-cell-like ability to produce many types of cells—and the breakdown of the cellular maturation process known as terminal differentiation," said senior author and MD Anderson President Ronald DePinho, M.D.
By searching for genes missing from GBM cells, rather than mutated, Hu and colleagues discovered a key differentiation pathway whose absence fuels tumor growth. "If glioblastoma cells were to undergo differentiation, the tumor would stop growing," Hu said. "But we've shown that if the terminal differentiation circuitry is gone, they get stuck in the middle and produce many different cell types."
Such cellular diversity, or heterogeneity, is a hallmark of cancer that helps it survive and progress. The "multiforme" in glioblastoma multiforme reflects the heterogeneity among and inside tumors.
The publication in PNAS is DePinho's Inaugural Paper, the first published in the journal by new members of the National Academy of Sciences. DePinho was elected to the prestigious academy in 2012. PNAS also published a question-and-answer interview with DePinho this week.
Cancer stem cells: heterogeneity machines
"When a normal neural stem cell divides, it makes one copy of itself and one copy of a precursor cell destined to differentiate into a neuron, an astrocyte or an oligodendrocyte," Hu said.
GBM cells appear locked in a stem-like state, which can lead to runaway division of undifferentiated cells.
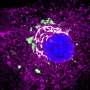
A microarray analysis of 71 human glioblastoma samples revealed high levels of stem cell and precursor cell markers for neurons and supportive cells. Fewer cells expressed markers of terminal differentiation. Overall, there were high levels of cellular heterogeneity dominated by immature cells. Higher-grade gliomas had greater heterogeneity.
Sifting genes involved in nervous system development
The team then surveyed The Cancer Genome Atlas GBM database looking for genes that have a known role in nervous system development and are frequently deleted. Of 71 genes identified, A2BP1 caused a notable reduction in colony formation in a GBM cell line. A2BP1 is a gene-splicing factor active in neural development that has been implicated in developmental and psychiatric disorders when mutated.
By profiling 430 TCGA GBM samples, the researchers found A2BP1 deleted in 10 percent of tumors. However, additional analysis showed that its protein is absent or steeply reduced in 90 percent of samples. The gene also is deleted in other nervous system tumors, and in 48 percent of colon cancer samples and 18 percent of sarcomas, suggesting a major tumor-suppressing role across cancers.
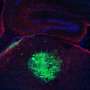
Silencing A2BP1 in GBM-prone premalignant neural stem cells led to tumor formation in mouse brains after 15 weeks, while control mice were tumor-free through 25 weeks. Forcing expression of the gene in mouse and human glioma stem cell lines impaired tumor formation by causing immature cells to try to differentiate into neurons, which subsequently died from apoptosis.
Gene that turns on A2BP1 identified
To explain the difference between A2BP1's deletion in only 10 percent of tumors but the absence or reduction of its protein in 90 percent of tumors, the team searched for transcription factors that turn on A2BP1 that might also be deleted or suppressed.
They found Myt1L deleted in 5 percent of samples and its protein absent or greatly reduced in 80 percent of tumors. Expression or suppression of Myt1L had similar effects in stem cell lines and mice to those caused by the same actions in A2BP1.
Myt1L also is one of three genes known to trans-differentiate fibroblast cells into neurons, this research makes the first connection between Myt1L and A2BP1, Hu said.
A multistep analysis of RNAs that interact with A2BP1 pointed to the known tumor-suppressor TPM1 as a key gene in mediating A2BP1's differentiation and cancer-blocking activity.
TPM1 proteins come in two forms, one found to have much higher cancer-blocking activity than the other. Splicing of TPM1 by A2BP1 increased levels of the version greater tumor-suppressing activity. Subsequent experiments showed that this version of TPM1 protein significantly reduced glioblastoma formation, invasion and migration in cell cultures and stymied tumor formation in mice.
A GBM-suppressing chain of events
In addition to discovering the deletion and suppression of A2BP1 in GBM, the team established that Myt1L switches on A2BP1, which splices TPM1 into a cell-differentiating, tumor-suppressing mode. "This is probably not the only way that A2BP1 suppresses tumors, but it's a key mechanism," Hu said.
Some therapies, mainly in blood malignancies, work by forcing immature cells to differentiate. There's been some hope that differentiation therapy might work on glioblastoma, but Hu notes that it's likely to be less effective if the cell's differentiation machinery is missing.
Their research could lead to biomarkers that indicate whether differentiation therapy will work against a given tumor. A combination of drugs that block stemness pathways and activate Myt1L-A2PB1 differentiation might provide an effective treatment for GBM, the authors noted.