Targeting cancer's sweet tooth
Ludwig researchers have elucidated a key mechanism by which cancer cells change how they metabolize glucose to generate the energy and raw materials required to sustain runaway growth.
Published online in Cell Metabolism, the Ludwig Cancer Research study also reveals how the aggressive brain cancer glioblastoma harnesses the mechanism to resist targeted therapies that should disrupt this capability—known as the Warburg effect—and suggests how such resistance might be overcome. In detailing the molecular circuitry of the phenomenon, the researchers uncover several possible targets for new drugs that might disrupt cancer cell metabolism to destroy tumors.
"Cancer and other fast-growing cells extract energy from glucose using a process that ordinarily kicks in only when oxygen is in short supply," explains Ludwig scientist Paul Mischel, MD, who is based at the University of California, San Diego School of Medicine. "This allows them to thread the needle: they get the energy they need from glucose but also retain the carbon-based building blocks for molecules like lipids, proteins and DNA, which dividing cells need in large quantities."
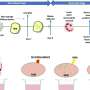
Until recently, relatively little was known about the biochemical circuits that induce this vital metabolic shift in cancer cells. Earlier this year, however, Mischel and his colleagues published a study describing how an aberrant growth signal found in many glioblastomas is channeled to induce the Warburg effect. That signaling cascade, which involves the key proteins PI3 kinase (PI3K), Akt and mTORC1, culminates in the activation of a transcription factor—a controller of gene expression—named c-Myc. "In many cancer cells," says Mischel, "c-Myc seems to be a lever that links growth signaling pathways with the machinery that controls the uptake and use of nutrients."
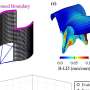
In the current study, Mischel, who did the research in collaboration with Ludwig researchers Kenta Masui, MD, PhD and Web Cavenee, PhD, both also at UC San Diego, identifies a second interacting biochemical cascade that is independent of the PI3K-Akt-mTORC1 signal and uses distinct biochemical circuits and an unusual mechanism to turn on c-Myc. This pathway, Mischel and his colleagues report, depends on signals from a protein complex named mTORC2. The researchers show that when mTORC2 is switched on, it silences two other transcription factors, FoxO1 and FoxO3, which would otherwise suppress the activation of c-Myc in the nucleus of the cell. Further, they learned that the silencing of the FoxOs occurs through a chemical modification—known as acetylation—a process that has not been well understood.
The study has significant implications for cancer therapy. "Many drugs have recently been devised to block PI3K-Akt-mTORC1 signaling," explains Mischel. "What we show is that when you use those drugs, you will probably drive the acetylation of the FoxOs through mTORC2, and inadvertently fuel the Warburg effect. In other words, this new pathway is likely to be responsible for resistance to those drugs. Our data suggest that to disrupt the Warburg effect and kill cancer cells, you have to develop therapies that target both signaling pathways. That's the main clinical ramification of this finding."
Mischel and his colleagues find that glioblastomas that rely predominantly on the mTORC2-mediated pathway tend to have the worse prognosis. Further, their studies suggest that lung cancer cells, too, use this pathway to induce the Warburg effect.
"Increasingly," says Mischel, "we're using glioblastoma as a system to understand a variety of other cancers and, in fact, this finding has broader relevance because the signaling pathways identified here are conserved across cancer types." Different cancers, he explains, are fueled by different types of mutations to growth factor receptors, but the signals these mutated receptors transmit tend to converge on a subset of signaling proteins.
"Our identification of the key molecules—and novel signaling mechanisms—involved in this pathway, has opened up a landscape rich in possible targets for novel cancer drugs," says Mischel. His laboratory, he says, is now working with other Ludwig researchers to identify small drug-like molecules that might disrupt key steps of the mTORC2-mediated pathway.