Better understanding of the HIV epidemic through an evolutionary perspective
With the abundance of sequencing data, scientists can use ever more powerful evolutionary biology tools to pinpoint the transmission and death rates for epidemics such as HIV, which has remained elusive to a cure. Reconstructed evolutionary trees, called phylogenies, can trace a family of viral mutations over time. When combined with epidemiology, tree construction can allow for great insight into the dynamics of disease transmission and how a pathogen eludes its host to spread infection.
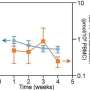
Professor Gabriel Leventhal, et. al., from the ETH Zurich in Switzerland report on a new method that successfully combines evolutionary tree studies and epidemiology, using viral sequence data from 10 transmission clusters of the Swiss HIV Cohort Study. For some clusters, the HIV epidemic appeared saturated, with very few new cases appearing while in others, new infections were still common. Overall, HIV transmission was characterized by initial rapid spread within subpopulations that slows down to only a small number of infections.
"Using a novel methodology, we were able to estimate the the number of individuals that are at risk of becoming infected within transmission clusters of the Swiss HIV epidemic and found that many of these clusters are characterized by initial rapid infection of most at risk individuals within a cluster, followed by a slowdown of new infections within each cluster," said Leventhal.
This allowed the team, for the first time, to estimate not only HIV transmission and death rates, but also the total susceptible population size within certain transmission groups from viral sequence data. Their model can successfully predict how the number of infected and susceptible individuals will vary over time, giving new insight and predictions into how an ongoing epidemic will continue to develop and help guide future public health intervention strategies.