Targeted investigational therapy potential to overcome crizotinib resistance in lung cancers
PF-06463922, an investigational drug being developed by Pfizer Inc., has the potential to become a new treatment option for patients who have lung cancer harboring abnormalities in the ALK gene, according to preclinical results presented at the AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics, held Oct. 19-23.
About 3 to 5 percent of lung cancers harbor ALK gene abnormalities. The drug crizotinib (Xalkori), which blocks ALK protein kinase activity, was approved in August 2011 by the U.S. Food and Drug Administration for the treatment of patients who have these lung cancers. Although robust responses to crizotinib are observed for lung cancers harboring ALK gene abnormalities, the majority eventually become resistant to the effects of the drug. In many cases, resistance arises because of genetic mutations in ALK.
"Resistance to targeted therapies such as crizotinib is a major challenge when treating patients with cancer," said Tod Smeal, Ph.D., associate research fellow in the Oncology Research Unit at Pfizer Inc. in San Diego, Calif. "Our goal is to take advantage of everything we have learned about designing drugs that target kinases like ALK and the ways in which lung cancers become resistant to crizotinib to develop the best next-generation ALK inhibitor we can.
"Our preclinical studies suggest that we are making progress toward achieving our goal: PF-06463922 has potent ALK-inhibiting activity, it is capable of inhibiting all the crizotinib-resistant ALK mutants so far detected in patients, and it can efficiently access the brain. We are excited about these preclinical results and very hopeful that they will translate into meaningful responses in the clinic."
After carefully designing PF-06463922, Smeal and colleagues first showed in cell assays that it potently inhibited the activity of ALK and all eight of the mutant forms of ALK known to cause resistance to crizotinib in patients with lung cancer. They then showed that PF-06463922 inhibited the growth of tumors harboring three of the crizotinib-resistant ALK mutants, including the most resistant ALK mutant, G1202R, in mice.
Further analysis indicated that PF-06463922 readily entered the brains of mice, rats, and dogs. In mice, levels of PF-06463922 in the brain were 20-30 percent of levels of PF-06463922 in the blood. This is potentially clinically relevant because a significant number of lung cancer patients will develop brain metastasis during the course of their disease, according to Smeal, although he noted that it will be important to see if these results in animals hold true in humans.
Smeal and colleagues also found that PF-06463922 potently inhibited the protein ROS1, a close relative of ALK recently implicated in a number of cancer types, including some lung and brain cancers. Further, PF-06463922 had antitumor effects in two mouse models of cancers driven by ROS1 gene abnormalities, leading the researchers to suggest that PF-06463922 has potential as a treatment for this subgroup of cancers, in addition to its promise as a new treatment for ALK-driven cancers.
More information:
Abstract Number: A277
Presenter: Tod Smeal, Ph.D.
Title: PF-06463922, a novel ROS1/ALK inhibitor, demonstrates sub-nanomolar potency against oncogenic ROS1 fusions and capable of blocking the resistant ROS1G2032R mutant in preclinical tumor models
Authors: Helen Y. Zou, Lars R. Engstrom, Qiuhua Li, Melissa West Lu, Ruth Wei Tang, Hui Wang, Konstantinos Tsaparikos, Sergei Timofeevski, Justine Lam, Shinji Yamazaki, Wenyue Hu, Hovhannes Gukasyan, Nathan Lee, Ted W. Johnson, Valeria Fantin, Tod Smeal. Pfizer, Inc., San Diego, CA
The oncogenic ROS1 gene fusion (Fig-ROS1) was first identified in glioblastoma cells over two decades ago. Recently, ROS1 gene rearrangements were further discovered in a variety of human cancers, including lung adenocarcinoma, cholangiocarcinoma, ovarian cancer, gastric adenocarcinoma, colorectal cancer, inflammatory myofibroblastic tumor, angiosarcoma, and epithelioid hemangioendothelioma, providing additional evidence for ROS1 as an attractive cancer target. The 1st generation Met/ALK/ROS1 inhibitor XALKORI ® (crizotinib) has demonstrated promising clinical response in ROS1 fusion positive NSCLC. But similar to what was seen with acquired ALK secondary resistant mutations in XALKORI refractory patients, a ROS1 kinase domain mutant—ROS1G2032R has been identified in a ROS1 positive NSCLC patient who developed resistance to XALKORI. Therefore, there is an urgent need to develop agents that can overcome this type of resistance.
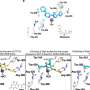
PF-06463922 is a novel, orally available, ATP-competitive small molecule inhibitor of ROS1/ALK with exquisite potency against ROS1 kinase. PF-06463922 inhibited the catalytic activity of recombinant ROS1 with a mean Ki of < 0.005 nM, and inhibited ROS1 autophosphorylation at IC50 values ranging from 0.1 nM to 1 nM cross a panel of cell lines harboring oncogenic ROS1 fusion variants including CD74-ROS1, SLC34A2-ROS1 and Fig-ROS1. PF-06463922 also inhibited cell proliferation and induced cell apoptosis at sub- to low-nanomolar concentrations in the HCC78 human NSCLC cells harboring SLC34A2-ROS1 fusions and the BaF3-CD74-ROS1 cells expressing human CD74-ROS1. In the BaF3 cells engineered to express the XALKORI resistant CD74-ROS1G2032R mutant, PF-06463922 demonstrated nanomolar potency against either ROS1G2032R cellular activity or cell proliferation. In vivo, PF-06463922 demonstrated marked cytoreductive antitumor efficacy at low nanomolar concentration in the NIH3T3 xenograft models expressing human CD74-ROS1 and Fig-ROS1. The antitumor efficacy of PF-06463922 was dose dependent and strongly correlated to inhibition in ROS1 phosphorylation and the downstream signaling molecules pSHP1, pSHP2 and pErk1/2, as well as inhibition of the cell cycle protein Cyclin D1 in tumors. To our knowledge, PF-06463922 is the first reported ROS1 inhibitor that is capable of blocking the resistant ROS1G2032R mutant at predicted pharmacologically relevant concentrations. Our data indicate that PF-06463922 has great potential for treating ROS1 fusion positive cancers including those from patients who relapsed from XALKORI therapy due to acquired ROS1G2032R mutation.
Abstract Number: PR10/B107
Presenter: Tod Smeal, Ph.D.
Title: Is CNS availability for oncology a no-brainer? Discovery of PF-06463922, a novel small molecule inhibitor of ALK/ROS1 with pre-clinical brain availability and broad spectrum potency against ALK-resistant mutations
Authors: Ted W. Johnson, Simon Bailey, Benjamin J. Burke, Michael R. Collins, J. Jean Cui, Judy Deal, Ya-Li Deng, Martin P. Edwards, Mingying He, Jacqui Hoffman, Robert L. Hoffman, Qinhua Huang, Robert S. Kania, Phuong Le, Michele McTigue, Cynthia L. Palmer, Paul F. Richardson, Neal W. Sach, Graham L. Smith, Lars Engstrom, Wenyue Hu, Hieu Lam, Justine L. Lam, Tod Smeal, Helen Y. Zou. Pfizer, Inc., San Diego, CA
Oncogenic fusions of Anaplastic Lymphoma Kinase (ALK) define a subset of human lung adenocarcinoma. The 1st generation ALK inhibitor crizotinib demonstrated impressive clinical benefit in ALK-fusion positive lung cancers and was approved by the FDA for the treatment of ALK-fusion positive NSCLC in 2011. However, as seen with most kinase inhibitors, patients treated with crizotinib eventually develop resistance to therapy. Acquired ALK kinase domain mutations and disease progression in the central nervous system (CNS) are reported as main contributors to patient relapse after ALK inhibitor therapy. Preclinically, crizotinib lacks significant brain penetration and does not potently inhibit activity of ALK kinase domain mutants, so a drug discovery program was initiated aimed to develop a second generation ALK inhibitor that is more potent than existing ALK inhibitors, capable of inhibiting the resistant ALK mutants and penetrating the blood-brain-barrier. These objectives present a considerable challenge in kinase inhibitor chemical space.
Here we report that PF-06463922, a novel small molecule ATP-competitive inhibitor of ALK/ROS1, showed exquisite potencies against non-mutant ALK (Ki <0.2 nM; cell IC50 ~2 nM) and ROS1 kinase (Ki <0.005 nM; cell IC50 ~0.2 nM), and demonstrated low nanomolar inhibitory activity against a panel of ALK kinase domain mutants representing all of the patient crizotinib resistant mutations reported to date. The more commonly reported L1196M gatekeeper mutant shows significant sensitivity to PF-06463922 (Ki 0.7 nM; cell IC50 16 nM). PF-06463922 is also very selective, and showed >100 fold kinase selectivity against 95% of the kinases tested in a 207 recombinant kinase panel.
Specific design considerations were developed leading to novel ATP-competitive kinase inhibitors with desired low efflux in cell lines over-expressing p-glycoprotein and breast cancer resistance protein, providing excellent blood-brain-barrier and cell penetration properties. Efforts to optimize ligand efficiency and lipophilic efficiency leveraging structure based drug design techniques led to ligands with overlapping broad spectrum potency and low efflux. Single and repeat dose preclinical rat in vivo studies of PF-06463922 demonstrated excellent oral bioavailability and CNS availability with free brain exposure approximately 30% of free plasma levels. In addition, CNS-directed safety studies showed no adverse events at predicted efficacious concentrations. It is anticipated that PF-06463922 with its potent activities on non-mutant ALK , ALK kinase domain mutations and CNS metastases would provide great promise for patients with ALK and ROS1 positive cancers.
Abstract Number: C253
Presenter: Tod Smeal, Ph.D.
Title: PF-06463922, a novel brain-penetrating small molecule inhibitor of ALK/ROS1 with potent activity against a broad spectrum of ALK resistant mutations in preclinical models in vitro and in vivo
Authors: Helen Y. Zou, Lars R. Engstrom, Qiuhua Li, Melissa West Lu, Ruth Wei Tang, Hui Wang, Konstantinos Tsaparikos, Jinwei Wang, Sergei Timofeevski, Dac M. Dinh, Hieu Lam, Justine Lam, Shinji Yamazaki, Wenyue Hu, Timothy Affolter, Patrick B. Lappin, Hovhannes Gukasyan, Nathan Lee, Jennifer M. Tursi, Ted W. Johnson, Valeria Fantin, Tod Smeal. Pfizer, Inc., San Diego, CA
Oncogenic fusions of Anaplastic Lymphoma Kinase (ALK) define a subset of human lung adenocarcinomas. The 1st generation ALK inhibitor XALKORI ® (crizotinib) demonstrated impressive clinical benefit in ALK-fusion positive lung cancers and was approved by the FDA for the treatment of ALK-fusion positive NSCLC in 2011. However, as seen with most kinase inhibitors, patients treated with XALKORI eventually developed resistance to therapy. Acquired ALK kinase domain mutations and brain metastases are significant contributors to the relapse after XALKORI therapy. To date, multiple types of ALK kinase domain mutations have been identified in XALKORI refractory patients including ALKG1269A, ALKL1196M, ALKC1156Y, ALKL1152R, ALKF1174L, ALKS1206Y, ALK1151Tins and ALKG1202R, accounting for about 1/3 of patient samples tested. Currently, a number of 2nd generation ALK inhibitors are under development aiming to overcome XALKORI resistant mutations. Even though in preclinical models, some ALK mutants such as ALKG1202R and ALK1151Tins confer high-levels of resistance to almost all of the 2nd generation ALK inhibitors tested.
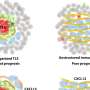
Here we report PF-06463922, a novel ATP competitive small molecule inhibitor of ALK/ROS1, with potent and selective inhibitory activity against all known acquired XALKORI resistant mutations identified in patients. PF-06463922 is also capable of penetrating the blood brain barrier in preclinical animal models. In vitro, PF-06463922 demonstrated potent inhibition in catalytic activities of ALK and 8 different ALK mutant kinases in recombinant enzyme and cell based assays (cell IC50s = 1 to 65 nM). PF-06463922 also showed potent growth inhibitory activity and induced apoptosis in the NSCLC cells harboring either non-mutant ALK or mutant ALK fusions (IC50s = 1 to 30 nM). In vivo, PF-06463922 demonstrated marked cytoreductive activity in mice bearing tumor xenografts that express EML4-ALK, EML4-ALKL1196M, EML4-ALKG1269A, EML4-ALKG1202R or NPM-ALK at low nM free plasma concentrations. These effects were associated with significant inhibition in cellular Ki67 and increased cleaved-caspase3 levels in tumors. In addition, PF-06463922 achieved brain exposure of 20-30% of its plasma levels in mice, and significantly regressed the brain tumors and prolonged survival of mice bearing orthotopic EML4-ALK and EML4-ALKL1196M positive brain tumor implants. The antitumor efficacy of PF-06463922 was dose dependent and strongly correlated with inhibition of ALK phosphorylation and downstream signaling. Our data indicate that PF-06463922 is the most potent ALK inhibitor reported to date (to our knowledge, against both non-mutant or mutant ALK in cell assays), and it demonstrates great potential for treating ALK fusion positive cancers including patients who relapsed from XALKORI therapy due to various ALK kinase domain mutations and/or brain metastases.