HRCT scans can identify deadly lung disease
People who have suspected idiopathic pulmonary fibrosis (IPF) without typical patterns on high resolution computed tomography scans could in future be spared the substantial risks of lung biopsy and be given a confident diagnosis of IPF based on clinical and radiological findings alone, according to new research published in The Lancet Respiratory Medicine.
Studies from the UK and USA suggest IPF is becoming more common. There are an estimated 5000 new cases in the UK each year and IPF causes more deaths each year than either leukaemia, ovarian cancer, or kidney cancer. In the USA, about 50 000 new cases are diagnosed each year and as many as 40 000 Americans die from IPF each year, the same number as die of breast cancer.
IPF causes progressive scarring of lung tissue, which eventually prevents the lungs from being able to supply the body with adequate oxygen. IPF has no cure and most people live only 3 to 5 years after diagnosis. Appropriate treatment is complicated by the fact that a definitive diagnosis often requires a lung biopsy.
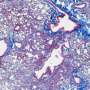
HRCT scans of the lungs can identify typical 'honeycombing pattern' of lung scarring and damage to the air sacs known as usual interstitial pneumonia (UIP) in people with IPF. In a patient with progressive breathlessness who has no significant environmental exposures attributable to pulmonary fibrosis, or evidence of collagen vascular diseases, the UIP pattern on HRCT is characteristic of IPF. When patients suspected to have IPF do not have the definitive UIP pattern on HRCT images, international guidelines recommend a surgical lung biopsy to make a confident diagnosis of IPF in such patients.
"Surgical lung biopsy is associated with substantial risks and many patients are too elderly, sick, and/or have comorbid conditions to tolerate the invasive procedure", explains Professor Ganesh Raghu from the University of Washington Medical Center, Seattle, USA who led the research.
"A confident diagnosis of IPF is needed to ensure that patients are well informed of the poor prognosis associated with IPF, are treated with the most appropriate therapies, consider participation in clinical trials of new therapies, and to identify those most suitable for lung transplantation."
In this retrospective study of 315 patients aged 40 years and older with little or no (5% or less) honeycombing on high resolution CT, 79 (94%) of 84 patients who had a high-resolution pattern of possible UIP diagnosed by an expert radiologist and pathologist had histopathological UIP confirmed after analysis of lung biopsy samples.
According to Raghu, "Our findings suggest that when a team of multidisciplinary experts in interstitial lung disease at a regional center (that includes a chest radiologist and a pulmonologist) work together to interpret possible UIP pattern on high-resolution CT in a patient suspected to have IPF, surgical lung biopsy might not be necessary to reach a diagnosis of IPF."
He concludes by cautioning, "Since the patients enrolled in our study were a highly selected cohort of patients suspected to have IPF and referred to regional sites for consideration of participation in a clinical trial, the findings from our study must not be extrapolated for all patients demonstrating the possible UIP pattern on HRCT images interpreted by general pulmonologists and radiologists in the community."
Writing in a linked Comment, Dr Simon Hart from Hull York Medical School in the UK says, "The results will also help to provide clarity about treatment options for a greater number of patients and increase the pool of patients suitable for inclusion in clinical trials of new therapies…In the modern era with optimal high-resolution CT imaging and thoracic radiologists familiar with interpretation of interstitial lung disease patterns through participation in multidisciplinary meetings, the role of lung biopsy assessment might diminish further. In future, combination of high-resolution CT with new noninvasive biomarkers and functional imaging could be used to better define phenotypes of fibrotic interstitial lung disease."
More information: Paper: www.thelancet.com/journals/lan … (14)70011-6/abstract