Quantifying genomic variation of herpes viruses is crucial step toward a vaccine

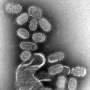
(Medical Xpress)—Unlike scientists studying the flu virus or HIV, researchers working to develop a vaccine for herpes simplex virus don't yet have a library containing thousands of sequenced viral genomes. Relative to the progress of flu- and HIV-related studies, herpes researchers are playing catch-up.
But a paper recently published in the Journal of Virology by Moriah Szpara and an international research team may help to advance the progress of herpes research across the globe. Szpara is assistant professor in biochemistry and molecular biology and holds a joint appointment in Penn State's Huck Institutes of the Life Sciences.
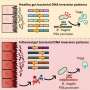
"In this paper," says Szpara, "we've compared for the first time 26 of the roughly 30 known herpes simplex virus genomes in order to gain some understanding of how different these viruses are around the planet."
"We need to know why some of them have much more virulence and greater lesion- and disease-severity outcomes," she says, "and also how much variation is out there that affects the virus's ability to escape from drugs and vaccine-based immunity. If we want to have a good vaccine, we need to know that we're priming the body's immune system with a target that's very stable in the virus, not an aspect the virus can easily escape from or that already has natural variants that differ from the vaccine target."
In quantifying the strains' genomic variation, the researchers also found a great deal of similarity (90 percent) between the viral proteins, "which," says Szpara, "is useful to know from a vaccine-design standpoint. There is hope that we can pick out proteins or regions of proteins that are relatively fixed or invariant in every strain we've looked at."
"There also turns out to be much more variation at the DNA level than we'd known before," she continues. "This tells us that there is definitely room for evolution in the virus and that we need to keep an eye on it. But it's not so much variation that we can't design good therapeutics or good vaccines – we can – it's just useful to know that it's not one precisely-matched strain found consistently around the world."
Unfortunately, the HSV strains that the team compared showed a number of mutations in their genomes which, according to Szpara, suggest that they had evolved in the lab since being isolated from their human hosts between roughly 25 and 80 years ago.
"Several of the strains couldn't have been in this state when they were isolated from human patients," she says, "since they're missing genes that are important for infection of animal models and, we presume, for infection of people, as well. Several viruses also have frame shifts in key proteins, suggesting that it's highly unlikely that these were the dominant viruses in the patients they came from."
Despite the considerable time that's passed since the strains were isolated, Szpara explains that the researchers have not been growing them constantly in a lab. "We can freeze and thaw them," she says, "so they may have spent on the order of days or months actively growing in the lab since they were taken."
"Essentially," she continues, "we've created a snapshot of the virus's variation – how different these 26 strains are from one another, and where variation happens within their genomes – but our snapshot's already old. I can't tell you what's circulating in Boston, New York City, San Francisco, Beijing, London, or anywhere else right now – nobody can. So while it's great that we now have this snapshot, we also have a tremendous imperative to fix this problem because humans are moving around the planet, and having sex and other intimate interactions, more often than ever before."
A simple kiss is enough to pass HSV-1 between individuals and intermix viruses, Szpara notes. Already the researchers have found among the viral strains included in the study one that doesn't seem to match the others' geographic pattern.
"Among these strains," Szpara explains, "when you try to cluster them to figure out who's more similar to whom, all but one virus strains clusters with its geographic neighbors. There was one strain from the U.S. that didn't cluster with the other ones from the U.S. Instead it went with the ones from Asia – and I think that's a forerunner of what you would find now. If you surveyed Boston now, you would find strains from all over the world, and I would guess that in any cosmopolitan city – and plenty of other places – where people from different geographic regions intermingle, there's been enough human movement and social interaction that these viruses are a little reflection of how friendly we've all become."
Now the researchers' task is to begin surveying viral strains in present-day populations and individuals, trying to determine how many different viruses people are shedding and how swiftly those change in their lifetimes.
"There are data suggesting that the viruses do change, and physicians and scientists have observed that patients can shed multiple viruses," Szpara explains. "How these viruses evolve in their human hosts is unclear, but it would really behoove us to figure this out. If current social trends are causing more virus exposures, that may impact virus evolution, and we need to know because it potentially changes how we think about drug and vaccine development."
More information: "Evolution and Diversity in Human Herpes Simplex Virus Genomes." Moriah L. Szpara, Derek Gatherer, Alejandro Ochoa, Benjamin Greenbaum, Aidan Dolan, Rory J. Bowden, Lynn W. Enquist, Matthieu Legendre, and Andrew J. Davison. J. Virol. January 2014 88:2 1209-1227; published ahead of print 13 November 2013, DOI: 10.1128/JVI.01987-13