Role of calcium in familial Alzheimer's disease clarified, pointing to new therapeutics
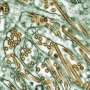
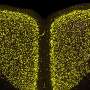
 and hyper-phosphorylated tau (antibody AT180) immunostaining of hippocampus from 18-month-old mice. Amyloid plaques (top row) and intracellular tau tangles (bottom row) in the 3xTg mouse were strongly reduced by genetic deletion of 50 percent of the IP3R1 in the 3xTg/Opt mouse. Wild-type (WT) and Opt mice expressing 50 percent of InsP3R exhibited no pathology. Credit: J. Kevin Foskett, Ph.D., & Dustin Shilling; Perelman School of Medicine at the University of Pennsylvania")
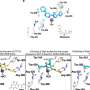
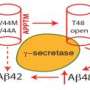
In 2008 researchers at the Perelman School of Medicine at the University of Pennsylvania showed that mutations in two proteins associated with familial Alzheimer's disease (FAD) disrupt the flow of calcium ions within neurons. The two proteins interact with a calcium release channel in an intracellular compartment. Mutant forms of these proteins that cause FAD, but not the normal proteins, result in exaggerated calcium signaling in the cell.
Now, the same team, led by J. Kevin Foskett, PhD, chair of Physiology, and a graduate student, Dustin Shilling, has found that suppressing the hyperactivity of the calcium channels alleviated FAD-like symptoms in mice models of the disease. Their findings appear this week in the Journal of Neuroscience.
Current therapies for Alzheimer's include drugs that treat the symptoms of cognitive loss and dementia, and drugs that address the pathology of Alzheimer's are experimental. These new observations suggest that approaches based on modulating calcium signaling could be explored, says Foskett.
The two proteins, called PS1 and PS2 (presenilin 1 and 2), interact with a calcium release channel, the inositol trisphosphate receptor (IP3R), in the endoplasmic reticulum. Mutant PS1 and PS2 increase the activity of the IP3R, in turn increasing calcium levels in the cell. "We set out to answer the question: Is increased calcium signaling, as a result of the presenilin-IP3R interaction, involved in the development of familial Alzheimer's disease symptoms, including dementia and cognitive deficits?" says Foskett. "And looking at the findings of these experiments, the answer is a resounding 'yes.'"
Robust Phenomenon
Exaggerated intracellular calcium signaling is a robust phenomenon seen in cells expressing FAD-causing mutant presenilins, in both human cells in culture and in mice. The team used two FAD mouse models to look for these connections. Specifically, they found that reducing the expression of IP3R1, the dominant form of this receptor in the brain, by 50 percent, normalized the exaggerated calcium signaling observed in neurons of the cortex and hippocampus in both mouse models.
In addition, using 3xTg mice – animals that contain presenilin 1 with an FAD mutation, as well as expressed mutant human tau protein and APP genes—the team observed that the reduced expression of IP3R1 profoundly decreased amyloid plaque accumulation in brain tissue and the hyperphosphorylation of tau protein, a biochemical hallmark of advanced Alzheimer's disease. Reduced expression of IP3R1 also rescued defective electrical signaling in the hippocampus, as well and memory deficits in the 3xTg mice, as measured by behavioral tests.
"Our results indicate that exaggerated calcium signaling, which is associated with presenilin mutations in familial Alzheimer's disease, is mediated by the IP3R and contributes to disease symptoms in animals," says Foskett. "Knowing this now, the IP3 signaling pathway could be considered a potential therapeutic target for patients harboring mutations in presenilins linked to AD."
The 'calcium dysregulation' hypothesis
"The 'calcium dysregulation' hypothesis for inherited, early-onset familial Alzheimer's disease has been suggested by previous research findings in the Foskett lab. Alzheimer's disease affects as many as 5 million Americans, 5 percent of whom have the familial form. The hallmark of the disease is the accumulation of tangles and plaques of amyloid beta protein in the brain.
"The 'amyloid hypothesis' that postulates that the primary defect is an accumulation of toxic amyloid in the brain has long been used to explain the cause of Alzheimer's", says Foskett. In his lab's 2008 Neuron study, cells that carried the disease-causing mutated form of PS1 showed increased processing of amyloid beta that depended on the interaction of the PS proteins with the IP3R. This observation links dysregulation of calcium inside cells with the production of amyloid, a characteristic feature in the brains of people with Alzheimer's disease.
Clinical trials for AD have largely been directed at reducing the amyloid burden in the brain. So far, says Foskett, these trials have failed to demonstrate therapeutic benefits. One idea is that the interventions started too late in the disease process. Accordingly, anti-amyloid clinical trials are now underway using asymptomatic FAD patients because it is known that they will eventually develop the disease, whereas predicting who will develop the common form of AD is much less certain.
"There has been an assumption that FAD is simply AD with an earlier, more aggressive onset," says Foskett. "However, we don't know if the etiology of FAD pathology is the same as that for common AD. So the relevance of our findings for understanding common AD is not clear. What's important, in my opinion, is to recognize that AD could be a spectrum of diseases that result in common end-stage pathologies. FAD might therefore be considered an orphan-disease, and it's important to find effective treatments, specifically for these patients - ones that target the IP3R and calcium signaling."