A new twist on neuro disease: Discovery could aid people with dystonia, Parkinson's and more
Twist and hold your neck to the left. Now down, and over to the right, until it hurts. Now imagine your neck – or arms or legs – randomly doing that on their own, without you controlling it.
That's a taste of what children and adults with a neurological condition called dystonia live with every day – uncontrollable twisting and stiffening of neck and limb muscles.
The mystery of why this happens, and what can prevent or treat it, has long puzzled doctors, who have struggled to help their suffering dystonia patients. Now, new re-search from a University of Michigan Medical School team may finally open the door to answering those questions and developing new options for patients.
In a new paper in the Journal of Clinical Investigation, the researchers describe new strains of mice they've developed that almost perfectly mimic a human form of the disease. They also detail new discoveries about the basic biology of dystonia, made from studying the mice.
They'll soon make the mice available for researchers everywhere to study, to accelerate understanding of all forms of dystonia and the search for better treatments. The lack of such mice has held back research on dystonia for years.
The U-M team's success in creating a mouse model for the disease came only after 17 years of stubborn, persistent effort – often in the face of setbacks and failure.
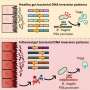
Led by U-M neurologist William Dauer, M.D., the team tried to figure out how and why a gene defect leads to an inherited form of dystonia that, intriguingly, doesn't start until the pre-teen or teen years, after which it progresses for many years but then stops getting worse after the person reaches their mid-20s.
The gene defect responsible, called DYT1, causes brain cells to make a less-active form of a protein called torsinA. But despite more than a decade of effort by Dauer's team and many others around the world, no one has been able to translate this information into an animal model with dystonia's characteristic movements.
Using the childhood onset as a clue, Dauer and his team used cutting-edge genetic technology to severely impair torsinA function during early brain development. This novel twist caused the new mice to closely mimic the human disease: they don't develop dystonia until they reach preteen age in "mouse years," and their symptoms stop getting worse after a while.
With this powerful tool in hand, Dauer's team were now able to peer into the brains of these animals to begin to unravel the mysteries of the disease.
In an unexpected development, they found that the lack of torsinA in the brains of dystonic mice led to the death of neurons – a process called neurodegeneration – in just a few highly localized parts of the brain that control movement. Like the dystonic movements, this neurodegeneration began in young mice, progressed for a time, and then became fixed.
"We've created a model for understanding why certain parts of the brain are more vulnerable to prob-lems from a certain genetic insult," says Dauer, an associate professor in the U-M departments of Neu-rology and Cell & Developmental Biology.
"In this case, we're showing that in dystonia, the lack of this particular protein during a critical window of time is causing cell death. Every disease is telling us something about biology—one just has to listen carefully."
More discoveries to come
Dauer and his team don't yet know why only one-third of human DYT1 gene mutation carriers develop primary dystonia during their school years, and why those who don't develop the disease before their early 20s will never go on to develop it.
They believe some critical events during the brain's development in infancy and childhood may have to do with it - and they're already working to explore that question in mice.
They also believe their mouse model will help them and other researchers understand how dystonia occurs in people who have Parkinson's disease, Huntington's disease, or damage caused by a stroke or brain injury. Some people develop dystonia without either a known gene defect or any of these other diagnoses – a condition called idiopathic dystonia.
In all these cases, as in people with DYT1 mutations, dystonia's twisting and curling motions likely arise from problems in the area of the brain that controls the body's motor control system.
In other words, something's going wrong in the process of sending signals to the nerves that control muscles involved in movement. Studying a "pure" form of dystonia using the mice will allow researchers to understand just what's going on.
The team's ultimate goal is to find new treatments for all kinds of dystonia. Currently, children, teens and young adults who develop it can take medications or even opt for a form of neurosurgery called deep brain stimulation. But the drugs carry major side effects and are only partially effective – and brain surgery carries its own risks. Dauer and his team are working to screen drug candidates.
More information: Journal of Clinical Investigation, Volume 124 Number 7, July 2014 DOI: 10.1172/JCI72830