Treating Machado Joseph Disease; a new approach to an old problem
Machado-Joseph disease (MJD) is a hereditary neurodegenerative disorder that destroys the brain areas involved in muscle control. Although the disease is clearly caused by a mutation in the ATXN3 gene resulting in an abnormal ataxin-3 protein that forms toxic aggregates in the brain, the mechanism of MJD development is unclear. And despite decades of research no cure or treatment has been found. But now, a study in the journal Brain by researchers from University of Coimbra in Portugal reports a new approach to solve this old problem, finding a treatment that can reverse the disease's neural damage and its symptoms in several animal models of MJD. The treatment restores normal levels of a molecule involved in protein regulation that is abnormally low in animals and human patients with MJD. Although much work needs to be done to see if this type of therapy could be applied to MJD patients, these are promising results, and the approach could be used in similar neurodegenerative diseases.
MJD (also known as spinocerebellar ataxia type 3 or SCA3) is rare, but can nevertheless show a remarkably high incidence (up to 1:150) in isolated populations. It is also very destructive. It is genetically transmitted, and every child of a sufferer has a 50 percent chance of developing the disease, with successive generations developing MJD earlier than the previous ones. Equally destructive is its progression, leaving the brain faculties intact while unremittingly destroying the body. Symptoms start with lack of coordination, speech and swallowing difficulties, and progress to various degrees of paralysis that can leave patients wheelchair-bound, totally dependent, or dead. The mutation first appeared in the small Azorean island of Flores, but has now spread all over the world, including to parts of the Australian aboriginal community where MJD is already considered as a serious problem. Despite extensive research, there is still no cure or treatment.
In an attempt to address the problem, Clévio Nóbrega, Pereira de Almeida and colleagues from the Center for Neuroscience and Cell Biology and the University of Pharmacy in Coimbra University, decided for once not to focus on ataxin-3, but instead on a protein called ataxin-2. It has already been suggested that ataxin-2 is abnormal in MJD, but it was only after ataxin-2 was recently discovered to regulate protein production that researchers started thinking that the protein could be important in MJD.
To investigate this possibility Nóbrega and colleagues looked for ataxin-2 in MJD patients' muscle cells and within post-mortem MJD brains. They found that, in fact ataxin-2 seemed to be largely absent in individuals with MJD, and what remained was found in the ataxin-3 toxic aggregates. Rodent models of the disease demonstrated that this reduction was driven by the increase in the toxic mutant ataxin-3 deposits as MJD progressed.
Excitingly, when the researchers restored ataxin-2 levels to normal in diseased animals, the toxic aggregates and the brain death typical of MJD started to disappear, with a corresponding reduction of disease symptoms. Since MJD patients have a similar lack of ataxin-2, these results suggest a potential new way to treat the disease in humans.
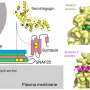
But how does ataxin-2 affect the production of the mutated ataxin-3 and MJD development? The answer, as Almeida's team discovered, was a molecule called PABP.
Proteins are the key molecules to all the body's reactions, and our DNA contains the instructions for their production. But to produce a new protein, the DNA information needs to be translated first into a molecule of RNA that can be "read" by our protein-producing factories, the ribosomes. PABP's job is to react with the RNA to help its translation, and without this reaction, the proteins cannot be produced. And ataxin-2 can bind PABP.
By blocking PABP or using a PABP molecule incapable of binding ataxin-2, the researchers were able to prove that this molecule was key to the effect of ataxin-2 in MJD.
In MJD animals and humans, ataxin-2 levels are very low, so there should be plenty of PABP available to push the production of the mutant protein. As a result, the disease fully progresses. But once Almeida and colleagues restore ataxin-2 levels to normal in diseased animals, PABP binds to it, halting ataxin-3 production. The toxic aggregates are constantly being cleared from the brain by the cell's "housekeeping" mechanisms, and once the production of the mutant protein is halted, the toxic aggregates and the neural damage start to disappear, along with the symptoms of the disease.
Although these new results look promising, the researchers note that much study needs to be done first to confirm these results and their clinical relevance.
More information: Clévio Nóbrega et al. Re-establishing ataxin-2 downregulates translation of mutant ataxin-3 and alleviates Machado–Joseph disease, Brain (2015). DOI: 10.1093/brain/awv298
Piece by Catarina Amorim