A new study identifies a previously unrecognized mutation that causes an inherited form of amyotrophic lateral sclerosis (ALS). The research, published by Cell Press in the December 9th issue of the journal Neuron, implicates defects in a cellular pathway linked with degradation of unwanted proteins in the underlying pathology of ALS and provides new insight into this incurable and fatal neurodegenerative disease.
ALS, also known as Lou Gehrig's disease, is a devastating disease that causes destruction of the neurons in the brain and spinal cord that control voluntary movement. There is no cure for ALS, which is characterized by a progressive paralysis that often leads to death from respiratory failure within three to five years of diagnosis. It is estimated that about 5% of ALS cases are inherited and a few genetic mutations linked with these familial cases of ALS have been identified.
"The identification of genes underlying rare familial forms of ALS has had a significant impact on our understanding of the molecular mechanisms underlying typical ALS," explains senior study author Dr. Bryan J. Traynor from the Laboratory of Neurogenetics at the National Institutes of Health in Bethesda, Maryland. "Each new gene implicated in the etiology of ALS provides fundamental insights into the pathogenesis of motor neuron degeneration, as well as facilitating disease modeling and the design and testing of targeted therapeutics; hence, there is much interest in the identification of novel genetic mutations."
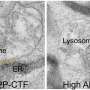
In an effort to further examine underlying genetic mutations associated with ALS, Dr. Traynor, along with his Italian collaborators Drs. Adriaon Chio, Gabriella Restagno and Jessica Mandrioli employed a sophisticated genetic screening technique to examine the entire "exome", all of the genes that carry instructions for making proteins, in a family with inherited ALS. Importantly, this particular ALS family did not exhibit mutations in genes previously associated with inherited ALS. The researchers identified a mutation in the gene for valosin-containing protein (VCP). VCP protein is part of the ubiquitin-proteasome machinery that degrades unwanted proteins inside the cell.
"Mutant VCP toxicity is partially mediated through its effect on a protein called TDP-43, a major constituent of the neuropathological inclusions that are characteristic of ALS and motor neuron degeneration," says Dr. Traynor. These findings validate the exome sequencing technique for identifying genetic causes of inherited ALS and are the first to implicate abnormalities in VCP and the cellular protein degradation pathway in ALS. "Our study potentially widens the clinical spectrum associated with ALS and provides new insight into this fatal disease," concludes Dr. Traynor.
Provided by Cell Press