Membrane molecule keeps nerve impulses hopping
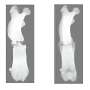
, Contactin-associated protein at the paranodes (blue) and potassium channels at the juxtaparanodes (red). Credit: Image source: Bhat lab, University of North Carolina at Chapel Hill.")
New research from the University of North Carolina at Chapel Hill School of Medicine describes a key molecular mechanism in nerve fibers that ensures the rapid conductance of nervous system impulses. The findings appear online Jan. 27, 2011 in the journal Neuron.
Our hard-wired nerve fibers or axons rely on an insulating membrane sheath, the myelin, made up of fatty white matter to accelerate the rate of transmission of electrical impulses from the brain to other parts of the body.
Myelin thus acts to prevent electrical current from leaking or prematurely leaving the axon. However, the myelin surrounding the axon isn't continuous; there are regularly spaced unmyelinated gaps about 1 micrometer wide along the axon. These unmyelinated regions named as nodes of Ranvier are where electrical impulses hop from one node to the next along the axon, at rates as fast as 160 meters per second (360 mph).
Determining exactly how the nodes of Ranvier function and how they are assembled, has fired the interest of neuroscientists for more than a century," said UNC neuroscientist Manzoor Bhat, PhD, Professor of Cell and Molecular Physiology in the UNC Neuroscience Research Center. "The answers may also provide important clues to the development of targeted treatments for multiple sclerosis and other disorders involving demyelination and/or disorganization of nodes of Ranvier."
Bhat and colleagues focused on a protein called Neurofascin 186, which accumulates in the membranes of axons at the nodes of Ranvier. Together with proteins Ankyrin-G and sodium channels, these molecules form a complex that facilitates passage of sodium ions through the channels in axons, thus making them paramount for the propagation of nerve impulses along myelinated nerve fibers.
Bhat's team had previously identified a homolog of Neurofascin in laboratory studies of Drosophila nerve fibers, and because its in vivo function had not been clearly defined in a mammalian system, they decided to study the function of this protein in laboratory mice.
Using targeted gene deletion methods, the UNC scientists genetically engineered mice lacking Neurofascin 186 in their neurons. "This caused the failure of sodium channels and Ankyrin-G to accumulate at the nodes of Ranvier. The result was paralysis, as there was no nerve impulse conductance," Bhat said.
According to Bhat, Neurofascin is an adhesion molecule that serves as the nodal organizer. "Its job is to cluster at the nodes of Ranvier. In doing so, it brings together sodium channels and Ankyrin-G where they interact to form the nodal complex. And if you don't have this protein, the node is compromised and there is no impulse propagation along the axon."
In further analysis, the researchers identified another important function of the nodes of Ranvier in myelinated nerve cells: to act as barriers to prevent the invasion of the nodal gap by neighboring paranodal molecular complexes. "So this tells us that sodium channels, Neurofascin 186, and Ankyrin-G must always remain in the node to have functional organization. If they don't, the flanking paranodes will move in and occupy the nodal gap and block nerve conduction," Bhat said.
The UNC neuroscientists see clinical implications for human disease. "In MS, for example, the proteins that make up the nodal complex start diffusing out from their normal location once you start losing the myelin sheath. If we can restore the nodal complex in nerve fibers, we may be able to restore some nerve conduction and function in affected axons." Their future studies are aimed at understanding whether the nodal complex could be reorganized and nerve conduction restored in genetically modified mutant mice.
"The discovery of an essential gap protein is exciting because it opens up the possibility that tweaking the protein could restore normal gap function in people with multiple sclerosis and other diseases in which the myelin sheaths and gaps deteriorate over time," said Laurie Tompkins, PhD, who oversees Manzoor Bhat's and other neurogenetics grants at the National Institutes of Health.