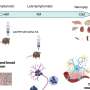
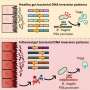
Based on a wide variety of genetic studies and analysis– from genome wide association studies looking for common variations in the DNA of many people with complex diseases to the sequencing of specific gene mutations thought to cause disease to whole genome sequencing – four Houston leaders in the field propose a unified genetic model for human disease.
"What emerges is a unified picture whereby previously distinct entities or categories of human diseases, chromosomal syndromes, genomic disorders, Mendelian disease, and multifactorial or complex traits, can now be considered as part of one continuum whereby common and rare variants including de novo (new) mutations in the context of environmental influences result in perturbation of the biological balance of a restricted set of networks activating final common pathways that ultimately cause disease," the authors wrote in an article that appears in the current issue of the journal Cell.
The authors include Dr. James Lupski, vice chair of molecular and human genetics at Baylor College of Medicine; Dr. Richard Gibbs, director of the Baylor Human Genome Sequencing Center; Dr. John Belmont, professor of molecular and human genetics at BCM, and Dr. Eric Boerwinkle, professor and director of the division of epidemiology at The University of Texas School of Public Health at Houston.
In other words, all kinds of genetic variation – changes in single genes (single nucleotide polymorphisms or SNPs), deletions or duplications of a large part of chromosomes (copy number variation), rare genetic variants, common variants – all play a role in a person's biologic continuum, health and risk of disease, said Lupski, also a professor of pediatrics.
"By trying to capture all variation rather than focusing on only one gene, one individual or one tool, you can understand the role of genetic variation in disease susceptibility more completely," said Lupski.
Using only one tool such as a genome-wide association study to look for common genetic variants in people with a disease such as high blood pressure or diabetes can bias the individual's perception of the problem, he said.
While genome-wide association studies have identified some common genetic variants that have a small effect on the risk of disease, the information is wanting for a more complete solution and is currently unable to be used for medical decision making.
As whole genome sequencing (determining the entire genetic make-up of a person) has come into play, it is apparent that people with many diseases have rare variants underlying them that have more effect on risk.
The rare variants are much more prominent than anticipated espoused the authors. Each genome sequenced has thousands of variants never seen before.
One thing that has become clear is that the variations inherited from your parents or their parents are more important to your risk of disease than those ancestral common variations identified in various populations.
"What genetic variation you inherited from your mom and dad and what arose in your recent relatives, as well as new mutations that arise in you, might be the most relevant for disease manifestation," said Lupski. "That's the concept of clan genomics."
"Considering only the population from which you came from is too broad a brush," said Gibbs. "Whereas looking at the recent generations can help you infer a lot about what your genetic susceptibilities might be."
While that may seem to be coming full circle in the notion of disease incidence, Gibbs insists. "We are not quite back where we started. The world around us has changed, and this paper is about that change."
"What is powerful are the beautiful examples (cited in the paper) of allelic (one form of a gene at a specific spot on the chromosome) interplay that give rise to subtle variations in disease," said Gibbs, who is also professor of molecular and human genetics at BCM.
The paper recounts studies that show that gene mutations that cause rare diseases can also contribute to more common, complex problems such as high blood pressure, high cholesterol and nonalcoholic fatty liver disease. Some genome-wide association studies have identified variants associated with common diseases that have a molecular cause that is similar to that found when mutated genes cause rare, inherited forms of the diseases.
Gibbs and Lupski said they hoped the paper encourages discussion about the best way forward in understanding disease and its genetic underpinnings.
"I'm more enthusiastic at looking in detail at a small number of cases and generalizing the genetics model from that rather than taking a superficial look at many cases and inferring from that,' said Gibbs.
"A couple of years ago, we were cataloguing the rare variants. Now we are saying that if we knit them together with an understanding of their functional role and the technology to bring genomics to the clinic, it will be time to hand these keys off to someone else," he said.
Provided by Baylor College of Medicine