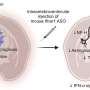
Professor MacLaren and Dr Zinkernagel perform gene therapy surgery for choroideraemia.
(Medical Xpress) -- The first patient to receive gene therapy for an incurable type of blindness was treated at the John Radcliffe Hospital in Oxford this week as part of a trial led by Oxford University.
If successful, the advance could lead to the first-ever treatment for choroideraemia, a progressive form of genetic blindness that first arises in childhood and is estimated to affect over 100,000 people worldwide.
‘This disease has been recognised as an incurable form of blindness since it was first described over a hundred years ago. I cannot describe the excitement in thinking that we have designed a genetic treatment that could potentially stop it in its tracks with one single injection,’ says Professor Robert MacLaren of the University of Oxford, who is leading the trial.
Jonathan Wyatt, 63, an arbitration lawyer from Bristol had the surgery at the Oxford Eye Hospital based at the John Radcliffe – the main NHS centre for this trial. He is the first of 12 people in this initial human trial that will receive the novel gene therapy.
Mr Wyatt was diagnosed with choroideraemia in his late teens and has suffered progressive sight loss ever since. He now sees only blackness except for a small area of a few degrees in diameter in the centre of his vision.
Choroideraemia is a genetic disease that leads to progressive degeneration of the retina in the eye. It generally affects males only and there is no treatment. The diagnosis is usually made in childhood and leads to blindness in men by their forties. It occurs due to deficiency of the REP1 gene located on the X chromosome.
The novel gene treatment was developed by Professor MacLaren at Oxford University in collaboration with Professor Miguel Seabra at Imperial College London. It is designed to provide the gene missing in people with choroideraemia to stop the deterioration that gradually leads to blindness.
It uses a virus essentially as a delivery vehicle that ferries DNA including the missing gene into the right part of the eye. The virus has been engineered to infect the light-sensitive cells in the retina known as photoreceptors. There the gene is switched on and becomes active.
With this particular gene therapy, the treatment could provide a one-off permanent correction of the disease because the gene is thought to remain in the retinal cells indefinitely.
‘This trial represents the world’s first ever attempt to treat this disease and the first time that gene therapy has been directed towards the light-sensitive photoreceptor cells of the human retina,’ says Professor MacLaren. ‘This represents a major breakthrough and is highly significant for patients who are losing sight from other photoreceptor diseases, such as retinitis pigmentosa.’
The trial will see 12 patients undergo surgery in which the gene therapy is injected into one eye. The other eye would then act as a control against which to assess any treatment effect. The researchers would however aim to go on to treat the second eye, should the treatment be proven to be effective.
The aim of the trial is primarily to assess safety, but it will also gain initial data on how effective the treatment is. The researchers estimate that it will take two years to know whether or not the degeneration has been stopped completely by the gene therapy.
‘While safety appears so far to be fine, the efficacy of the gene therapy will only be evident after 24 months. We need this time to measure any effect as the degeneration caused by choroideraemia is slow,’ explains Professor MacLaren, who is also an honorary consultant at the Oxford Eye Hospital and Moorfields Eye Hospital.
The clinical trial is funded by a grant awarded to the University of Oxford by the Health Innovation Challenge Fund – a translational award scheme funded jointly by the Wellcome Trust and the Department of Health.
Professor Seabra, who played a key role at Imperial College London in identifying the gene causing choroideraemia and in eliciting the mechanism of cell death in the retina, comments: ‘The ability to offer a gene replacement treatment for these patients was the final objective of 20 years of intense research in my laboratory. This is a moment of fulfilment for us and a dream come true for all choroideraemia patients.’
Provided by Oxford University