Blocking telomerase kills cancer cells but provokes resistance, progression
Inhibiting telomerase, an enzyme that rescues malignant cells from destruction by extending the protective caps on the ends of chromosomes, kills tumor cells but also triggers resistance pathways that allow cancer to survive and spread, scientists report in the Feb. 17 issue of Cell.
"Telomerase is overexpressed in many advanced cancers, but assessing its potential as a therapeutic target requires us to understand what it does and how it does it," said senior author Ronald DePinho, M.D., president of The University of Texas MD Anderson Cancer Center.
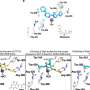
"We exploited the experimental merits of mice to model and study more precisely telomere crisis, telomerase reactivation and telomerase extinction in cancer development, progression and treatment," DePinho said. "This elegant model exposed two mechanisms, including one unexpected metabolic pathway, used by cancer cells to adapt to loss of telomerase.
"These findings allow us to anticipate how tumor cells might respond to telomerase inhibition and highlight the need to develop drug combinations that target telomerase and these adaptive resistance mechanisms," DePinho said.
Researchers evaluated telomerase as a therapeutic target in experiments that originated in DePinho's lab at the Dana-Farber Cancer Institute in Boston. He became MD Anderson's fourth full-time president in September.
Telomerase activity is low or absent in normal cells, which have segments of repeat nucleotides called telomeres at the ends of their chromosomes that protect DNA stability during cell division, said first author Jian Hu, Ph.D., an instructor in MD Anderson's Department of Cancer Biology.
With each division the telomeres shorten, leading eventually to genomic instability and cell death, a period termed "telomere crisis," Hu said. In cancer, telomerase becomes active during telomere crisis and rescues the genomically abnormal cells by lengthening telomeres.
In a series of experiments in a lymphoma mouse model, the team found:
- Telomerase reactivation in malignant cells after genomic instability causes cancer progression.
- Inhibiting telomerase caused tumor cell death but also led to alternative lengthening of telomeres (ALT) independent of telomerase.
- ALT-positive cells increase both the expression and copy number of a gene called PGC-1ß, a key regulator of mitochondrial function, to compensate for mitochondrial and reactive oxygen species defense deficiencies.
- Targeting PGC-1ß to weaken mitochondria function enhances anti-telomerase therapy.
Telomerase reactivation causes aggressive cancer
Third- and fourth-generation mice with telomerase activated by 4-OHT had a median survival of 30 days and more frequent tumor infiltration to the spleen, kidney, liver, lung, bone marrow and brain than did control-treated mice, 70 percent of which lived beyond 50 days. Tumor cells in control-treated mice were more likely to be detected and destroyed by tumor-suppressing p53 signaling.
"These findings are consistent with telomere crisis leading to genomic instability during early stage cancer, with reactivated telomerase protecting malignant cells later to ensure tumor progression," Hu said.
Telomere dysfunction causes cancer-promoting genetic changes
Later-generation mice with activated telomerase had 4,928 amplified genes and 2,297 deletions. The team compared these changes to those in human lymphoma tumors and found 565 matching amplified genes and 300 deletions.
These cross-species copy number alterations included several known tumor-suppressing genes and oncogenes, suggesting that initial telomere dysfunction not only drives primary tumor development but also confers malignant traits such as invasiveness.
Telomerase extinction works - at first
The team then took tumor cells from late-generation mice with activated telomerase - the aggressive tumors - and passaged them four times through groups of mice treated with either 4-OHT to trigger telomerase production or the control vehicle that leaves the enzyme off.
During the first two rounds, survival for the two groups was about the same. In the third round, the control mice had a major improvement in survival over the telomerase arm, indicating that telomere erosion had allowed cellular defense mechanisms to pick off genomically unstable cells.
However, in the fourth passage, survival of the control-treated mice fell back toward that of mice with active telomerase. The tumors had become resistant without relying on telomerase.
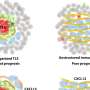
Alternative lengthening of telomeres rescues cancer cells
An analysis showed that telomere lengths of tumor cells with active telomerase remained largely unchanged across the four passages. Telomeres shortened in cells lacking telomerase through the first two passages followed by a sharp increase during the third and fourth passages.
Other molecular evidence pointed to alternative lengthening of telomeres in telomerase-absent cells.
They found that ALT-positive tumors had different gene expression patterns - 891 genes with increased expression, 1,345 with decreased - compared to telomerase-positive tumors.
Key gene in mitochondrial pathway active in ALT cells
Many genes were found in networks regulating mitochondrial biology and oxidative stress regulation. PGC-1ß was the only gene in both pathways with increased expression and copy number gain in the ALT-positive tumors.
PGC-1ß is a master regulator of both pathways, which turn out to be dysfunctional in ALT-positive tumors. When the researchers knocked down PGC-1ß, mice with ALT-positive tumors survived much longer than those with intact PGC-1ß and than those with activated telomerase in their tumors.
In normal cells, power-generating mitochondria process fatty acids to produce ATP, a molecule that serves as the major energy source for the cell. Cancer cells generally rely more on sugar processing to generate energy. However, DePinho and colleagues note their genetic evidence suggests that mitochondria play a role supporting cancer cells.