New proteins to clear the airways in cystic fibrosis and COPD
University of North Carolina scientists have uncovered a new strategy that may one day help people with cystic fibrosis and chronic obstructive pulmonary disorder better clear the thick and sticky mucus that clogs their lungs and leads to life-threatening infections. In a new report appearing online in The FASEB Journal, researchers show that the "SPLUNC1" protein and its derivative peptides may be able to help thin this thick mucus by affecting the epithelial sodium channel (ENaC). Not only does this research have implications for cystic fibrosis and COPD, but it also enhances the understanding of hypertension due to the role it also plays in controlling blood pressure.
"We hope that this study will pave the way for a new class of peptide-based channel inhibitors that can help reverse the mucus dehydration seen in Cystic Fibrosis and COPD," said Robert Tarran, Ph.D., a researcher involved in the work from the Cystic Fibrosis/Pulmonary Research and Treatment Center at the University of North Carolina in Chapel Hill. "This would help restore mucus clearance and kick-start the lung's ability to clear unwanted pathogens."
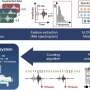
To identify which part of SPLUNC1 actually affects ENaC, scientists eliminated parts of the protein until it lost function. In fact, even after the eliminating 85 percent of SPLUNC1, it still affected ENaC, suggesting that the ENaC inhibitory domain was in the remaining 15 percent. Researchers then synthesized an 18-amino acid peptide of this region and tested its ability to bind to ENaC and to inhibit fluid absorption in human bronchial epithelial cells derived from people with and without cystic fibrosis. This peptide inhibited ENaC and fluid absorption in all systems tested, without affecting structurally-related ion channels. They also found that ENaC activity was affected for more than 24 hours in cystic fibrosis airway cultures, suggesting that this peptide may be therapeutically beneficial for the treatment of cystic fibrosis patients who suffer from over-active ENaC and consequentially have too little lung fluid.
"Breathing is something most healthy people take for granted." said Gerald Weissmann, M.D., Editor-in-Chief of The FASEB Journal. "However, people with cystic fibrosis and COPD battle for every breath because sticky mucus plugs their airways. This research should give scientists a new way of clearing the air for people with cystic fibrosis and COPD."
More information: Carey A. Hobbs, Maxime G. Blanchard, Stephan Kellenberger, Sompop Bencharit, Rui Cao, Mehmet Kesimer, William G. Walton, Matthew R. Redinbo, M. Jackson Stutts, and Robert Tarran. Identification of SPLUNC1's ENaC-inhibitory domain yields novel strategies to treat sodium hyperabsorption in cystic fibrosis airways. FASEB J, doi:10.1096/fj.12-207431