Researchers identifie gatekeeper protein, new details on cell's power source
Researchers at Temple University's Center for Translational Medicine and the University of Pennsylvania have identified a protein that serves as a gatekeeper for controlling the rush of calcium into the cell's power source, the mitochondria. Without this calcium spigot under control, calcium levels can run amok, contributing to cardiovascular disease, diabetes and neurodegeneration. The findings, reported online October 25, 2012, in the journal Cell, add important new insights into the inner workings of the mitochondria and may eventually help scientists better understand and target certain cellular processes gone awry, leading to new therapies for disease.
"Calcium is crucial for cell signaling, and keeping calcium at a certain level in the mitochondria is important to help regulate various cell processes and physiology," said co-senior author Muniswamy Madesh, PhD, Assistant Professor of Biochemistry at Temple University School of Medicine and a member of Temple's Center for Translational Medicine. "We've shown this gatekeeper establishes a threshold for calcium and prevents it from rushing in and overwhelming the mitochondria, which if unregulated, can act as a sponge and soak up large amounts of calcium in the cell. These results may help us find new ways to control calcium levels and head off problems that might lead to disease."
Maintaining a proper level of calcium is imperative for cells to work properly, and is particularly important in the mitochondria. Cells rely on mitochondria to generate usable energy sources in the form of the chemical ATP, which is necessary to carry out normal cellular metabolism and other activities. ATP production in turn depends on calcium, or rather, charged calcium ions that can flow into the mitochondria from the cell's reservoir in the cytoplasm. Scientists have studied calcium uptake by mitochondria for some five decades, but the details of the mechanisms for managing it under normal conditions were unclear.
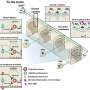
Dr. Madesh, co-senior author Kevin Foskett, PhD, at the University of Pennsylvania, and their co-workers may have at least in part solved this mystery. They found a molecular mechanism – a mitochondrial "gatekeeper" protein called MICU1 – that guards a protein pore, controlling how much calcium comes into the mitochondria. The researchers found that MICU1 works with this calcium channel pore, MCU, to set a threshold for the amount of calcium coming into the mitochondria specifically to enable the cell to maintain a level of calcium in mitochondria under normal "resting conditions."
Using a technique to silence gene and protein expression, the researchers found that when they turned off MICU1, excess cellular calcium was rapidly taken up by the mitochondria. When they re-expressed the molecule, they found that once again the calcium influx was under control. MICU1 detected calcium in the surrounding mitochondrial matrix at a certain level, maintaining comparatively low levels of mitochondrial calcium – about five to six times lower than what is considered "equilibrium."
"This gave us a clue that maybe there is a threshold at which mitochondria sense cellular calcium, and this protein acts like a sensor," Dr. Madesh noted.
"We've shown that the MICU1 establishes and controls a set point, which is crucial to maintaining the cell's calcium homeostasis," Dr. Madesh said. "Mitochondria in healthy cells rely on this mechanism to protect from calcium overload under physiological conditions. Disrupting this gatekeeper and the set point and chronically elevating mitochondrial calcium could lead to damage in neurons, and in the heart, liver, and other organs. Mitochondrial calcium is important for metabolic and cardiovascular functions, and maintaining this homeostasis is crucial. Cells lacking the set point will lead to mitochondrial dysfunction and cell death."
The findings suggest an array of potential therapeutic options to explore, including gene therapy, said first author Karthik Mallilankaraman, PhD, a postdoctoral fellow in the Department of Biochemistry and the Center for Translational Medicine at Temple University School of Medicine.