The same genetic defect causes Pompe disease in both humans and dogs

Pompe disease, a severe glycogen storage disease appearing in Lapphunds is caused by a genetic defect in acid α-glucosidase gene. The same genetic mutation also causes the equivalent disease in humans. Based on this finding, canine Pompe disease can now be diagnosed with a genetic test.
This research was completed at the Canine Genetics Research Group lead by professor Hannes Lohi in the University of Helsinki and Folkhälsan Research Center in Finland and will be published in PLOS ONE on February 14, 2013.
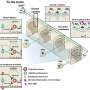
Human Pompe disease is caused by complete or partial deficiency of the acidic α-glucosidase enzyme. In humans, over 300 mutations have been discovered in the gene encoding this enzyme. Pompe disease is recessively inherited, therefore requiring the inheritance of the defected gene from both father and mother. Typically, human patients have two different mutations; one is inherited from father and the other from mother. If both mutations cause severe defect to the enzyme, the enzymatic activity is completely lost. This leads to infantile onset Pompe disease.
The average onset of the infantile disease is before two months of age and the affected children often die before the age of one year. Typical symptoms are floppy appearance, difficulties in sucking and swallowing, an enlarged heart which may lead to heart failure, difficulties in breathing, and respiratory infections. There is no cure for the disease, but enzyme replacement therapy relieves the symptoms and prolongs the patient's lifespan.
Canine Pompe disease is known to occur in Swedish Lapphunds and in this study it was revealed also from Finnish Lapphunds. Both the affected puppies and infantile onset patients lack functional acid α-glucosidase enzyme. Normally this enzyme breaks down the stored glycogen into glucose inside the lysosomes. When the α-glucosidase enzyme is not working properly, glycogen accumulates inside the cells of all tissues. In Pompe disease, glycogen doesn't break down naturally and accumulation most severely affects tissue function of the muscles.
Fast discovery of the gene defect
"From previous studies we knew which enzyme is defective in human patients, so we had a clear candidate gene for our canine studies" states professor Hannes Lohi, the leader of the Canine Genetics Group.
Sequencing the gene from the Finnish Lapphund litter revealed a recessively inherited mutation. This mutation is the same as one of the known 300 mutations in humans. Affected dogs carried two copies of the mutation, the parents were carriers and healthy siblings were either carriers or free of the mutation.
"The effect of the mutation to the gene product is so severe, that although we only had two affected cases in our study, we are certain we have identified the causative mutation" assures the first author of the paper Dr. Eija Seppälä.
The research was carried out in collaboration with Dr. Arnold Reuser who works in the Erasmus MC, Netherlands. With Dr. Reusers' help, a stored cell sample from an affected Swedish Lapphund born already in the 1970s was also tested. This dog was affected by the same mutation as the Finnish Lapphunds.
Finnish Lapphund, Swedish Lapphund and Lapponian Herder have common ancestors from the original reindeer-herding dog population. Therefore one aim was to discover the mutation frequency among current Lapphund and Lapponian Herder populations. A carrier frequency of 5% among Finnish Lapphunds and 2% among Lapponian Herders was revealed by testing approximately 100 dogs from both breeds. In addition, a total of 34 Swedish Lapphunds from Finland, Sweden and Norway were tested. All of these dogs were healthy and did not carry the mutation. It is possible that the sample size was too small for Swedish Lapphund breed and therefore carriers were not detected or it is possible that rigorous breeding program in the 1980s and 1990s has eliminated all the carriers from the current breed.