Accused of complicity in Alzheimer's, amyloid proteins may be getting a bad rap
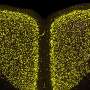
Amyloids—clumps of misfolded proteins found in the brains of people with Alzheimer's disease and other neurodegenerative disorders—are the quintessential bad boys of neurobiology. They're thought to muck up the seamless workings of the neurons responsible for memory and movement, and researchers around the world have devoted themselves to devising ways of blocking their production or accumulation in humans.
But now a pair of recent research studies from the Stanford University School of Medicine sets a solid course toward rehabilitating the reputation of the proteins that form these amyloid tangles, or plaques. In the process, they appear poised to turn the field of neurobiology on its head.
The first study, published in August, showed that an amyloid-forming protein called beta amyloid, which is strongly implicated in Alzheimer's disease, could reverse the symptoms of a multiple-sclerosis-like neurodegenerative disease in laboratory mice.
The second study, to be published April 3 in Science Translational Medicine, extends the finding to show that small portions of several notorious amyloid-forming proteins (including well-known culprits like tau and prion proteins) can also quickly alleviate symptoms in mice with the condition—despite the fact that the fragments can and do form the long tendrils, or fibrils, previously thought harmful to nerve health.
"What we're finding is that, at least under certain circumstances, these amyloid peptides actually help the brain," said Lawrence Steinman, MD, professor of neurology and neurological sciences and of pediatrics. "This really turns the 'amyloid-is-bad' dogma upside down. It will require a shift in people's fundamental beliefs about neurodegeneration and diseases like multiple sclerosis, Alzheimer's and Parkinson's."
Steinman is a noted expert in multiple sclerosis whose research led to the development of natalizumab (marketed as Tysabri), a potent treatment for the disease.
Taken together, the studies begin to suggest the radical new idea that full-length, amyloid-forming proteins may in fact be produced by the body as a protective, rather than destructive, force. In particular, Steinman's study shows that these proteins may function as molecular chaperones, escorting and removing from sites of injury specific molecules involved in inflammation and inappropriate immune responses.
Steinman, who is also the medical school's George A. Zimmermann Professor, is the corresponding author of the research. Jonathan Rothbard, PhD, a senior research scientist in the Steinman laboratory, is the senior author; postdoctoral scholar Michael Kurnellas, PhD, is the lead author.
Although the specific findings of Steinman's two studies are surprising, there have been inklings from previous research that amyloid-forming proteins may not be all bad. In particular, inhibiting, or knocking out, the expression of several of the proteins in the mouse models of multiple sclerosis—a technique that should block the course of the disease if these proteins are the cause—instead worsened the animals' symptoms.
And there's the fact that these so-called dangerous amyloid-forming molecules are surprisingly prevalent. "We know the body makes a lot of amyloid-forming proteins in response to injury," said Steinman. "I'm doubtful that that's done to produce more harm. For example, the prion protein is found in every cell in our bodies. What is it doing? It's possible that any therapeutic maneuver to remove all of these proteins could interfere with their natural function."
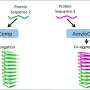
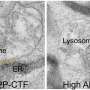
Understanding how amyloids form requires an understanding of the biology of proteins, which are essentially strings of smaller components called amino acids attached end to end. Once they're made, these protein strings twist and fold into specific three-dimensional shapes that fit together like keys and locks to do the work of the cell.
A misfolded protein is likely to be unable to carry out its duties and must be disposed of by the body's cellular waste-management system. Amyloid-forming proteins (of which there are around 20), however, don't go quietly, if at all. Instead, they initiate a chain reaction with other misfolded proteins—forming long, insoluble strands called fibrils that mat together to form amyloid clumps. These clumps appear consistently in the brains of people with neurodegenerative diseases like Alzheimer's and multiple sclerosis, but not in the brains of healthy people.
Although these clumps are thought to be detrimental to nerve cells, it's not entirely clear how they cause harm. One possibility is the ability of the fibrils to form cylindrical pores that could disrupt the cellular membrane and interfere with the orderly flow of ions and molecules used by the cells to communicate and transmit nerve signals. Regardless, their very presence suggests a diagnosis of neurodegeneration to many clinicians, including—until recently—Steinman.
"We began this research because these molecules are present in the brains of people with multiple sclerosis," said Steinman. "We expected to show that the presence of beta amyloid made the disease worse in laboratory animals. Instead, we saw a great deal of benefit."
Intrigued by the results of their first study, the researchers next tested the effect of small, six-amino-acid portions of several amyloid-forming proteins, including beta amyloid, which appeared likely to share a three-dimensional structure. They found that nearly all of the tiny protein molecules, or hexamers, were also able to temporarily reverse the symptoms of multiple sclerosis in the mice (when the treatment was stopped, the mice developed signs of the condition within a few days).
The researchers noted, however, that the curative effect of the hexamers was linked to their ability to form fibrils similar, but not identical, to their longer parent molecules. For example, these simplified hexamer fibrils are more easily formed and broken apart than those composed of whole proteins. They are also thought not to be able to form the cylindrical pores that might damage cell membranes. Finally, the hexamer fibrils appear to inhibit the formation of fibrils from full-length proteins—perhaps by blocking, or failing to promote, the chain reaction that initiates fibril formation.
When Steinman and his colleagues mixed the fibril-forming hexamers with blood plasma from three people with multiple sclerosis, they found that the fibrils bound to and removed from solution many potentially damaging molecules involved in inflammation and the immune response.
"These hexamer fibrils appear to be working to remove dangerous chemicals from the vicinity of the injury," said Steinman.
The researchers are eager to pursue the use of these small hexamers as therapies for neurodegenerative diseases like multiple sclerosis. Much research is still needed, but Steinman is hopeful.
"The lessons we learn from our study of amyloid-forming proteins in multiple sclerosis could be helpful for stroke and brain trauma, as well as for Alzheimer's," said Steinman. "We're gaining insight into how current therapeutic approaches may be affecting the body, and beginning to understand the nuances necessary to design a successful treatment. Although it will take time, we're determined to move promising results out of the laboratory and into the clinic as quickly as possible."