Structure that edits messenger RNA transcripts defective in two different forms of motor neuron diseases
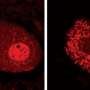
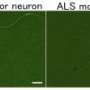
 TDP-43 (green) and SMN (red) proteins localize in nuclear gems in cultured cells (arrows). (Lower) Spliceosome subunits accumulate abnormally in the nuclei of motor neurons from an ALS patient (right), but not in normal motor neurons (left). Credit: 2013 K. Yamanaka et al.")
Amyotrophic lateral sclerosis (ALS) and spinal muscular atrophy (SMA) are degenerative motor neuron diseases in which the key mutated genes are involved in RNA metabolism. This similarity suggests that a common dysregulation of some aspect of RNA metabolism in motor neurons may underlie both disorders, although the exact cellular effects of the neurodegenerative mutations are unknown. Koji Yamanaka, Hitomi Tsuiji and colleagues from the RIKEN Brain Science Institute and other institutions in Japan have now obtained evidence that a cellular structure that edits messenger RNA (mRNA) transcripts is defective in both of these motor neuron diseases.
ALS is associated with mutations in the SOD1, TDP-43 and FUS/TLS protein-encoding genes, and spinal muscular atrophy (SMA) with mutations in a gene called SMN1. Identifying that the TDP-43, FUS/TLS and SMN proteins are all localized to structures known as 'gems' inside the nucleus, the researchers investigated whether these proteins might perform a similar function, which may indicate that the associated diseases share common RNA processing defects.
Yamanaka and his colleagues performed a series of biochemical experiments using lab-grown neurons and cancer cells, as well as neurons isolated from genetically engineered mice lacking the FUS gene. They found that eliminating TDP-43 expression in cultured cells prevented the formation of nuclear gems, and that gems were absent from neurons isolated from the mutant mice. Gem formation requires an interaction between TDP-43, SMN and FUS proteins, and this interaction is mediated by one end of the TDP-43 protein.
The researchers also found that all three proteins are involved in maintenance of the spliceosome, a large multi-component structure found in the nucleus. The spliceosome consists of multiple protein and RNA subunits, and controls splicing—the process by which non-coding sequences are removed from mRNA transcripts before they are translated into the strings of amino acids that make up proteins.
In ALS patients, the team found that ALS motor neurons had spliceosome defects—gems were missing from the nucleus and the RNA subunits of the spliceosome accumulated abnormally. Motor neurons from SMA patients, on the other hand, had significantly reduced levels of spliceosome RNA subunits in the nucleus.
The findings suggest that loss of spliceosome integrity plays an important role in neurodegeneration in both diseases. "Defective spliceosomes cause abnormal protein expression patterns, which can lead to motor neuron death," says Yamanaka. "This could be a new therapeutic target for neurodegenerative diseases, and we are now initiating efforts to develop a new class of drugs."
More information: Tsuiji, H. et al. Spliceosome integrity is defective in the motor neuron diseases ALS and SMA. EMBO Molecular Medicine 5, 221–234 (2013). dx.doi.org/10.1002/emmm.201202303