How serotonin receptors can shape drug effects, from LSD to migraine medication

New findings by researchers carrying out experiments at the U.S. Department of Energy Office of Science's Advanced Photon Source (APS) help explain why some drugs that interact with two kinds of human serotonin receptors have had unexpectedly complex and sometimes harmful effects.
"Understanding the structure-function of these receptors allows us to discover new biology of serotonin signaling and also gives us better ideas about what biological questions to probe in a more intelligent manner," said The Scripps Research Institute (TSRI) Professor Raymond Stevens, who was a senior investigator for the new research. The studies were published in two papers that appeared in the May 3, 2013, issue of the journal Science.
Stevens's laboratory at TSRI has pioneered the development of techniques for determining the three-dimensional atomic structures of cellular receptors—particularly the large receptor class known as G protein-coupled receptors (GPCRs). GPCRs sit in the cell membrane and sense various molecules outside cells. When certain molecules bind to them, the receptors respond in a way to transmit a signal inside the cell.
"Because G protein-coupled receptors are the targets of nearly 50 percent of medicines, they are the focus of several major National Institutes of Health (NIH) initiatives," said Jean Chin of the NIH's National Institute of General Medical Sciences (NIGMS), which partly funded the work through the Protein Structure Initiative. "These detailed molecular structures of two serotonin receptor subfamilies bound to antimigraines, antipsychotics, antidepressants or appetite suppressants will help us understand how normal cellular signaling is affected by these drugs and will offer a valuable framework for designing safer and more effective medicines."
In the past several years, using x-ray crystallography, the Stevens laboratory has determined the high-resolution structures of 10 of the most important GPCRs for human health—including the β2 adrenergic receptor, the A2a adenosine receptor (the target of caffeine); the human immunodeficiency virus-related CXCR4 receptor; the pain-mediating nociceptin receptor; the S1P1 receptor important for inflammatory diseases; the H1 histamine receptor (antihistamine medications); and the D3 dopamine receptor which is involved in mood, motivation, and addiction.
Serotonin receptors are no less important. "Nearly all psychiatric drugs affect serotonin receptors to some extent, and these receptors also mediate a host of effects outside the brain, for example on blood coagulation, smooth muscle contraction, and heart valve growth," said Bryan Roth, a collaborator on both studies, who is a professor of pharmacology at the University of North Carolina (UNC).
Roth's laboratory teamed up with Stevens's as part of the NIGMS Protein Structure Initiative. For this project the two labs also worked with the laboratories of Professors Eric Xu and Hualiang Jiang at the Shanghai Institute of Materia Medica, part of the Chinese Academy of Sciences. "By collaborating with the Chinese teams we were able to complete a much more thorough study and get the most out of our fundamental structural results," said Stevens.
In the first of the new studies, co-lead author Chong Wang, a graduate student in the Stevens laboratory, and his colleagues determined the structure of the serotonin receptor subtype 5-HT1B, the principal target of several drug classes. (5-HT, or 5-hydroxytryptamine, is a technical term for serotonin.) The team produced the 5-HT1B receptor while it was bound by either ergotamine or dihydroergotamine—two old-line anti-migraine drugs that work in part by activating 5-HT1B receptors.
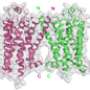
With the help of the special fusion protein, nicknamed BRIL (apocytochrome b562RIL), Wang and colleagues were able to stabilize these structures and coax them to line up in a regular ordering known as a crystal. X-ray crystallography carried out at the General Medical Sciences and Cancer Institutes Structural Biology Facility (GM/CA-XSD) 23-ID-D and Life Sciences Collaborative Access Team 21-ID-D x-ray beamlines at the APS at Argonne National Laboratory revealed, at high-resolution, an atomic structure of 5-HT1B with a main binding pocket and a separate, extended binding pocket.
In the second study, TSRI graduate student and lead author Daniel Wacker and colleagues used similar techniques at GM/CA-XSD beamline 23-ID-D to determine the structure of the 5-HT2B receptor bound to ergotamine. The 5-HT2B receptor was chiefly of interest because drug developers want to avoid activating it.
"Drugs that are meant to target other serotonin receptors in the brain can have harmful off-target effects on 5-HT2B receptors, which are found abundantly on heart valves, for example," said Roth. The weight-loss drug fenfluramine and closely related dexfenfluramine were withdrawn from the US market in 1997 after being linked to heart valve disease. Roth's laboratory later showed that this side effect was mediated by heart valve 5-HT2B receptors.
Analyses of the 5-HT1B and 5-HT2B receptor structures revealed a subtle difference between them. "Although their main binding pockets look very similar, their extended binding pockets are not as similar—the one for 5-HT2B is narrower and in a slightly different position," said Wang.
With the two receptor structures in hand, the Xu and Jiang team simulated the bindings of various drugs. They showed, for example, that anti-migraine drugs called triptans should bind well to 5-HT1B receptors but poorly to 5-HT2B receptor structures, in which the extended binding pocket is less accessible. Similarly, the team's calculations confirmed that fenfluramine's active metabolite should bind very tightly to the 5-HT2B receptor.
In the second study, the researchers used the 5-HT2B and 5-HT1B structural data to better understand a recently discovered GPCR signaling pathway.
When a neurotransmitter such as serotonin binds to its GPCR receptor and triggers the primary, G protein-mediated activation signal, it also usually triggers another signal, often mediated by a protein called β-arrestin. This second signaling cascade may simply have the effect of "arresting" or inhibiting the primary, G protein-mediated signaling. But it can also have other effects on the cell, and although most molecules bind to their target GPCRs in a way that activates these primary and secondary signals equally, others preferentially activate one or the other.
"Such functional selectivity, as we call it, adds another layer of complexity to drug effects on GPCRs," said Roth, a co-senior author of the study.
Roth's laboratory produced several 5-HT receptor subtypes in test cells, and compared the strength of G-protein and β-arrestin signaling when these receptors were bound by ergotamine or various other drugs, including the ergotamine-derived hallucinogen LSD (lysergic acid diethylamide). Most of the tested drugs showed no bias. However, ergotamine, LSD and some of their relatives turned out to be clearly biased in favor of β-arrestin signaling at the 5-HT2B receptor. Comparison of the ergotamine-bound 5-HT2B structure with the ergotamine-bound 5-HT1B structure revealed the likely reason.
"We could see that when ergotamine is bound to the 5-HT2B receptor it stabilizes the receptor structure in a conformation that interferes with G protein signaling," said Wacker.
The findings allow scientists to start probing this arrestin-mediated signaling pathway and its downstream effects in a more targeted manner.
"These structural data are teaching us to ask better questions about receptor biology," said Stevens.
More information: Wang, C. et al. Structural Basis for Molecular Recognition at Serotonin Receptors, Science 340, 610 (3 May 2013). DOI: 10.1126/science.1232807