For a healthy brain, don't let the trash pile up

Recycling is not only good for the environment, it's good for the brain. A study using rat cells indicates that quickly clearing out defective proteins in the brain may prevent loss of brain cells.
Results of a study in Nature Chemical Biology suggest that the speed at which damaged proteins are cleared from neurons may affect cell survival and may explain why some cells are targeted for death in neurodegenerative disorders. The research was supported by the National Institute of Neurological Disorders and Stroke (NINDS), part of the National Institutes of Health.
One of the mysteries surrounding neurodegenerative diseases is why some nerve cells are marked for destruction whereas their neighbors are spared. It is especially puzzling because the protein thought to be responsible for cell death is found throughout the brain in many of these diseases, yet only certain brain areas or cell types are affected.
In Huntington's disease and many other neurodegenerative disorders, proteins that are misfolded (have abnormal shapes), accumulate inside and around neurons and are thought to damage and kill nearby brain cells. Normally, cells sense the presence of malformed proteins and clear them away before they do any damage. This is regulated by a process called proteostasis, which the cell uses to control protein levels and quality.
In the study, Andrey S. Tsvetkov and his colleagues from the University of California, San Francisco (UCSF) and Duke University, Durham, N.C., showed that differences in the rate of proteostasis may be the clue to understanding why certain nerve cells die in Huntington's, a genetic brain disorder that leads to uncontrolled movements and death.
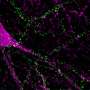
To measure how quickly proteins are cleared away from cells, the researchers developed a new technique called optical pulse-labeling, allowing them to follow specific proteins in individual living cells. To test the technique, they grew brain cells in a dish and turned on Dendra2, a photoswitchable protein that glows from green to red after being hit by a specific type of light. Both the red and green glow can be followed until the protein is cleared from the cell. In this way, the researchers could track the lifetime of newly produced Dendra2 (which glows green) and older, photoswitched Dendra2 (which glows red) until the protein was cleared away from the cell.
"Before this new technique, there was no way to look at individual neurons and their capacity to handle proteins. This method provides a real-time readout of how fast proteins are turned over in neurons and gives us a look at some of the mechanisms involved," said Margaret Sutherland, Ph.D., program director at NINDS.
The researchers followed Dendra2 in a set of striatal neurons, which they obtained from rats. The striatum (where striatal neurons are located) is a brain region involved in a number of brain functions including planning movements and is most heavily affected in Huntington's disease. They discovered that the mean lifetime of the protein (how long it remained in the cell) varied three- to fourfold, suggesting that rates of proteostasis were different among individual neurons. In other words, some cells may process an identical protein much slower than others.
Then, the researchers investigated how cells deal with different forms of huntingtin, the protein involved in Huntington's. They fused Dendra2 on the end of a normal or mutant version of huntingtin to track how long the protein remained in cells. The mutant version of huntingtin is longer, and contains three building blocks of the protein repeated an abnormal number of times. These repeats in huntingtin are what cause it to misfold, eventually leading to neuron death and the symptoms of the disease. As predicted, in their experiments, the mutant form of huntingtin caused more rat cells to die than did the normal form of the protein.
The researchers found that the amount of time the mutant protein remained in the cell predicted neuronal survival: shorter mean lifetimes of mutant huntingtin were associated with longer neuronal survival. A shorter mean lifetime indicates that a protein does not remain in the cell for a long time, and that proteostasis is working effectively to clear it away. This suggests that improving proteostasis in Huntington's brains may improve neuronal survival.
To test this idea, the researchers activated Nrf2, a protein known to regulate protein processing. When Nrf2 was turned on, the mean lifetime of huntingtin was shortened, and the neuron lived longer.
"Nrf2 seems like a potentially exciting therapeutic target. It is profoundly neuroprotective in our Huntington's model and it accelerates the clearance of mutant huntingtin," said Dr. Steven Finkbeiner, senior author of the paper.
Although both striatal and cortical neurons are affected by mutant huntingtin, striatal neurons are more susceptible to cell death. The investigators found that striatal neurons were not as effective as cortical neurons in recognizing and clearing away the mutant protein.
"One surprising finding from these experiments was the significance of single cells' ability to clear mutant huntingtin. It turned out that this ability largely predicted their susceptibility, whether that neuron came from the most vulnerable region of the brain – the striatum, or the cortex, which is less vulnerable," said Dr. Finkbeiner. The findings indicate that the toxicity of the damaged proteins may cause neurodegeneration by interfering with the proteostasis system, affecting how quickly they are cleared from neurons.
"The results should remind us that focusing on the disease-causing proteins is only one side of the coin. To understand why some cells die and others are spared, we may need to recognize that there are major, largely unrecognized cell-specific differences in the ways that various types of neurons recognize and dispose of disease-causing proteins," continued Dr. Finkbeiner.
The researchers explored potential mechanisms behind differences in proteostasis. One way that cells normally get rid of proteins is through autophagy—a process in which proteins are packed up into spheres and then broken down. Results in this paper suggested that neurons increased the rate of autophagy when they sensed that the mutant form of huntingtin was accumulating, indicating the autophagy system may be a drug target.
"These findings provide evidence that our brains have powerful coping mechanisms to deal with disease-causing proteins. The fact that some of these diseases don't cause symptoms we can detect until the fourth or fifth decade of life, even when the gene has been present since birth, suggests that those mechanisms are pretty good," said Dr. Finkbeiner.
Future research is needed to determine why coping mechanisms fail as brain cells age and how neurons in the healthy brain keep the proteostasis system functioning.
"New research methods that help us understand how individual neurons function will increase our understanding of central nervous system disorders and help identify new treatments. It is critical to continue working on the methods such as those described in this paper," said Dr. Sutherland.
More information: Nature Chemical Biology, July 21, 2013 DOI: 10.1038/nchembio.1308