Neuroscientists identify protein linked to Alzheimer's-like afflictions
A team of neuroscientists has identified a modification to a protein in laboratory mice linked to conditions associated with Alzheimer's Disease. Their findings, which appear in the journal Nature Neuroscience, also point to a potential therapeutic intervention for alleviating memory-related disorders.
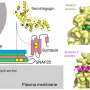
The research centered on eukaryotic initiation factor 2 alpha (eIF2alpha) and two enzymes that modify it with a phosphate group; this type of modification is termed phosphorylation. The phosphorylation of eIF2alpha, which decreases protein synthesis, was previously found at elevated levels in both humans diagnosed with Alzheimer's and in Alzheimer's Disease (AD) model mice.
"These results implicate the improper regulation of this protein in Alzheimer's-like afflictions and offer new guidance in developing remedies to address the disease," said Eric Klann, a professor in New York University's Center for Neural Science and the study's senior author.
The study's co-authors also included: Douglas Cavener, a professor of biology at Pennsylvania State University; Clarisse Bourbon, Evelina Gatti, and Philippe Pierre of Université de la Méditerranée in Marseille, France; and NYU researchers Tao Ma, Mimi A. Trinh, and Alyse J. Wexler.
It has been known for decades that triggering new protein synthesis is vital to the formation of long-term memories as well as for long-lasting synaptic plasticity—the ability of the neurons to change the collective strength of their connections with other neurons. Learning and memory are widely believed to result from changes in synaptic strength.
In recent years, researchers have found that both humans with Alzheimer's Disease and AD model mice have relatively high levels of eIF2alpha phosphorylation. But the relationship between this characteristic and AD-related afflictions was unknown.
Klann and his colleagues hypothesized that abnormally high levels of eIF2alpha phosphorylation could become detrimental because, ultimately, protein synthesis would diminish, thereby undermining the ability to form long-term memories.
To explore this question, the researchers examined the neurological impact of two enzymes that phosphorylate eIF2alpha, kinases termed PERK and GCN2, in different populations of AD model mice—all of which expressed genetic mutations akin to those carried by humans with AD. These were: AD model mice; AD model mice that lacked PERK; and AD model mice that lacked GCN2.
Specifically, they looked at eIF2alpha phosphorylation and the regulation of protein synthesis in the mice's hippocampus region—the part of the brain responsible for the retrieval of old memories and the encoding of new ones. They then compared these levels with those of postmortem human AD patients.
Here, they found both increased levels of phosphorylated eIF2alpha in the hippocampus of both AD patients and the AD model mice. Moreover, in conjunction with these results, they found decreased protein synthesis, known to be required for long-term potentiation—a form of long-lasting synaptic plasticity—and for long-term memory.
To test potential remedies, the researchers examined phosphorylation of eIF2alpha in mice lacking PERK, hypothesizing that removal of this kinase would return protein synthesis to normal levels. As predicted, mice lacking PERK had levels of phosphorylated eIF2alpha and protein synthesis similar to those of normal mice.
They then conducted spatial memory tests in which the mice needed to navigate a series of mazes. Here, the AD model mice lacking PERK were able to successfully maneuver through the mazes at rates achieved by normal mice. By contrast, the other AD model mice lagged significantly in performing these tasks.
The researchers replicated these procedures on AD model mice lacking GCN2. The results here were consistent with those of the AD model mice lacking PERK, demonstrating that removal of both kinases diminished memory deficits associated with Alzheimer's Disease.
More information: Suppression of eIF2a kinases alleviates Alzheimer's disease–related plasticity and memory deficits, DOI: 10.1038/nn.3486