Autism gene stunts neurons, but growth can be restored, in mice
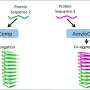
 and a less branched neuron with the NHE6 defect (right). Neurons below (no defect on the left, with defect on the right) received BDNF. Credit: Morrow lab/Brown University")
Brown University researchers have traced a genetic deficiency implicated in autism in humans to specific molecular and cellular consequences that cause clear deficits in mice in how well neurons can grow the intricate branches that allow them to connect to brain circuits. The researchers also show in their study (online Sep. 12, 2013, in Neuron) that they could restore proper neuronal growth by compensating for the errant molecular mechanisms they identified.
The study involves the gene that produces a protein called NHE6. Mutation of the gene is directly associated with a rare and severe autism-related condition known as Christianson syndrome. But scientists, including senior author Dr. Eric Morrow, have also associated the protein with more general autism.
"In generalized autism this protein is downregulated," said Morrow, assistant professor of biology in the Department of Molecular Biology, Cellular Biology, and Biochemistry at Brown and a psychiatrist who sees autism patients at the Bradley Hospital in East Providence. "That meant to us that downregulation of NHE6 is relevant to a sizeable subset of autism."
The NHE6 protein helps to regulate acidity in the endosomes of cells. These endosomes are responsible for transporting material around cells and for degrading proteins including ones that signal neurons to grow the elaborately branched axons and dendrites that form neural connections.
In their experiments the researchers measured acidity in the endosomes of brain cells of normal mice and in mice with mutations in the NHE6 gene. They found that the mutant mice had significantly higher endosome acidity. The mutant mice with the higher endosome acidity also had more degradation of a receptor protein, called TrkB, that responds a neurotrophic factor called BDNF. Together they signal axon and dendrite growth and branching.
Did the higher acidity and lower levels of TrkB signaling affect the neurons? Morrow and his colleagues were able to show directly in the mouse brain that the neuronal branching was diminished as were the number and maturity of connections between neurons, called synapses. Further still, working with co-author Julie Kauer, professor of medical science in the Department of Molecular Pharmacology, Physiology, and Biotechnology, they looked at synaptic and circuit function in the mice, and they found deficits corresponding to those anatomical findings.
"One of the overriding problems in disorders like autism, we think, is that it's a problem of communication between different areas of the brain and neurons communicating with each other in networks," said Morrow, who is affiliated with the Brown Institute for Brain Science.
Searching for a rescue
Having discovered a specific chain of events by which NHE6 mutations undermine neural branching and connectivity, Morrow and lead authors Qing Ouyang and Sofia Lizarraga sought to find out why and whether they could fix it.
Sometimes acidity in the endosome can activate protein-degrading enzymes called proteases. The team hypothesized that perhaps the acidity resulting from the absence of NHE6 was leading proteases to degrade TrkB, reducing its levels in mutant neurons compared to normal ones. When they treated mutant cells with a protease inhibitor called leupeptin, they found that the TrkB levels and signaling returned to levels close to those found in the normal cells.
Given that TrkB's job is to bind with BDNF, the researchers also hypothesized that if the problem of NHE6 mutation was a reduction of TrkB, perhaps a suitable end-run around the problem would be to administer BDNF to cells directly. Indeed they found that NHE6 mutant cells, if given extra BDNF, produced axon and dendrite growth and branching that was more like normal neurons.
"In this paper we show that BDNF signaling is attenuated in the mutant mice, but it's not blocked," Morrow said. "You can rescue the [neuronal growth] by turning up the signaling."
There are already drugs developed to deliver doses of chemicals that increase or mimic BDNF in the body, Morrow said, but many more tests beyond this study would have to be done before scientists and doctors could know whether a BDNF-related drug could have a therapeutic effect for patients with Christianson syndrome or any related form of autism.
"We don't think that this is everything about the condition," Morrow said. "But if we were able to treat this one mechanism by adding exogenous drug, would it repair enough or some element of it?"
Christianson syndrome and perhaps only a subset of autism appears to relate to deficits in neural branching. Some forms of autism, in fact, may result from too much branch growth. Moreover, doctors have no precise ways to tell whether a child diagnosed with autism has too much or too little neural branching.
But given the study results suggesting that NHE6 may play a role in some autism forms perhaps by hindering neural branching, the new research suggests a target for addressing it.