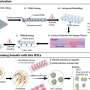
A mouse model predicted that inhibition of the enzyme MEK using targeted therapy may provide an effective, single-agent treatment for pediatric patients with neurofibroma, and early results from a phase I trial support this finding, according to the data presented here at the American Association for Cancer Research (AACR) special conference on "Pediatric Cancer at the Crossroads: Translating Discovery Into Improved Outcomes," held Nov. 3-6.
"We found that MEK inhibition shrank most neurofibromas in a genetically engineered mouse model of neurofibromatosis type 1," said Nancy Ratner, Ph.D., Beatrice C. Lampkin chair of cancer biology at Cincinnati Children's Hospital Medical Center. "Scientists are hopeful that genetically engineered mouse models can be used to identify therapies that will work in human patients."
Neurofibromas are benign tumors growing in the peripheral nerves that affect about 95 percent of people with the inherited disease neurofibromatosis type 1. About 25 to 50 percent of affected children develop a subtype of this disease, plexiform neurofibroma, which can become malignant. Neurofibromas are fueled initially by the mutation of the tumor-suppressing gene NF1. The NF1 protein normally functions to deactivate Ras-GTPase cell signaling, but treatments aimed at blocking Ras signaling have not led to effective therapies. The most effective therapies found in neurofibroma mouse models to date are drug candidates that target inhibition of MEK, a protein involved in Ras signaling.
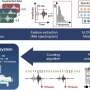
"Supporting the possibility that blocking the Ras-MEK-ERK pathway is a molecular target for treating neurofibromatosis type 1-driven disease, physicians at the National Cancer Institute (NCI) found that five out of nine neurofibromatosis type 1 subjects with plexiform neurofibromas treated with a MEK inhibitor showed partial responses," Ratner said.
In an NCI-coordinated phase I study, patients were treated with selumetinib (AZD6244), an orally bioavailable inhibitor of MEK1/2. Patients experiencing a partial response had their tumor volumes shrink by 20 percent or more. The researchers found that tumor shrinkage occurred slowly over time in patients with progressive and nonprogressive plexiform neurofibroma, and progressive disease has not been observed to date after a median treatment time of 10 months.
Based on these encouraging results, enrollment in the clinical study is ongoing, and a phase II study is being planned, according to Ratner.
Previous preclinical studies by Ratner and colleagues showed effectiveness of MEK inhibitors in a genetically engineered mouse model of plexiform neurofibroma. They administered the MEK inhibitor PD-0325901 in this mouse model at doses ranging from 0.5 to 10 mg/kg per day for two months, and found that neurofibromas shrank at all administered dose levels. In addition, they found that administering the MEK inhibitor prior to neurofibroma growth caused a size reduction in the tumors.
Because mutations of the NF1 gene are frequently found in other cancers including glioblastoma, lung adenocarcinoma, and ovarian cancer, this work may be relevant to other tumor types as well, according to Ratner.
The preclinical studies were funded by the Children's Tumor Foundation Neurofibromatosis Therapeutic Consortium. The phase I clinical trial is sponsored by the NCI Cancer Therapy Evaluation Program and funded, in part, by the NCI Center for Cancer Research's intramural research program and by a Clinical Trial Award from the Children's Tumor Foundation. Ratner has declared no conflicts of interest.
More information: www.aacr.org/page34138.aspx
Provided by American Association for Cancer Research