Uncovering first molecular missteps that drive neurons in pathway leading to Alzheimer's disease
Much of the debate in Alzheimer's disease (AD) has focused on whether the protein amyloid-beta or the tau protein is the symptom or the cause of the disease.
But George Bloom, Ph.D., and colleagues at the University of Virginia (UVA) have pursued another hypothesis: much earlier interactions between the two proteins them drive adult neurons into the forbidden pathway of "cell cycle re-entry" (CCR).
Dr. Bloom will present additional evidence supporting the role of CCR in AD on Sunday, Dec. 15, at the American Society for Cell Biology (ASCB) annual meeting in New Orleans. He will report the identification of a critical balance point between tau and a master cellular regulator that amyloid-beta oligomers disrupt.
A novel group of proteins, Rac1, Gαs (Gs alpha) and NCAM, and two protein kinase complexes, mTORC1 and mTORC2, play a role in driving mature neurons to their death, according to the UVA researchers.
These new findings are scientific sequels to a series of dramatic discoveries by the Dr. Bloom's lab that unravel the molecular events that drive adult neurons in CCR. As Dr. Bloom noted in his research presentation at last year's ASCB annual meeting, most normal adult neurons are supposed to be permanently post-mitotic—that is, they have finished dividing and are locked out of the cell cycle.
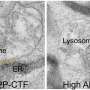
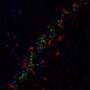
Yet in AD, neurons frequently re-enter the cell cycle, fail to complete mitosis and ultimately die. In late stage AD, up to 30 percent of the neurons in the frontal lobes of the brain are dead, surrounded by large amyloid plaques and tau-associated neurofibrillary tangles.
Dr. Bloom said that he views AD as fundamentally a problem of the cell cycle, with both amyloid-beta and tau required for the interaction that pushes neurons into destructive CCR. "The massive neuron death that occurs in AD therefore appears to be caused by the raw ingredients of plaques and tangles working in concert with each other, rather than by the plaques and tangles themselves," Dr. Bloom explained.
This new research to be presented at ASCB 2013 follows up on results published last spring when the UVA researchers described in greater detail how amyloid-beta activates multiple enzymes named protein kinases to add phosphates to specific sites on tau, setting neurons on the pathway to CCR.
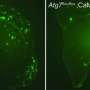
Now in this new molecular "sequel," Dr. Bloom and lab members Andrés Norambuena and Lloyd McMahon implicate a novel group of proteins—Rac1, Gαs (Gs alpha), and NCAM—and two protein kinases complexes—mTORC1 and mTORC2—as required participants to set off CCR.
Their identification reveals how a fundamental balance is upset, placing neurons on the road to AD. "The mTOR complexes are master regulators of cellular proliferation, growth and metabolism," Dr. Bloom said. "Most important, our results indicate that tau normally inhibits mTOR from promoting neuronal cell replication, but that this inhibition is reversed by an amyloid beta oligomer-induced, mTOR-dependent mechanism that modifies tau. In other words, tau and mTOR regulate each other."
This delicate balance is compromised by amyloid-beta oligomers in a way that allows neurons that should never replicate to re-enter the cell cycle. They fail to divide and eventually die instead. "Some of the earliest events in AD pathogenesis are therefore caused by amyloid-beta oligomers altering a fundamental neuronal signaling axis centered around tau and mTOR," proposed Dr. Bloom who said that he believes that the proteins identified in this signaling axis are potential biomarkers, and therapeutic targets for very early stages of AD, leaving the door open for an even more exciting sequel down the road.
More information: Author will present, "Ectopic Cell Cycle Re-entry, a Major Cause of Neuron Death in Alzheimer's Disease Is Controlled by Rac1 and mTOR," Sunday, Dec. 15, 2013, during the 12 noon to 1:30 p.m. poster session, "Rho-Family GTPases."