Mechanism discovered for how amyotrophic lateral sclerosis mutations damage nerve function
St. Jude Children's Research Hospital scientists led a study showing that mutations in a gene responsible for amyotrophic lateral sclerosis (ALS) disrupt the RNA transport system in nerve cells. The findings appear in the current issue of the scientific journal Neuron and offer a new focus for efforts to develop effective treatments.
The findings offer a new avenue for researchers to pursue in the quest for desperately needed treatments for ALS, a disorder that kills most patients within five years of diagnosis. ALS, also known as Lou Gehrig's disease, is diagnosed in about 5,600 individuals nationwide each year and is associated with muscle weakness and paralysis.
The gene, TDP-43, carries instructions for making a protein of the same name. While mutations in TDP-43 were known to cause ALS and a related neurodegenerative disorder, until now the mechanism involved was a mystery. This study showed for the first time that the mutations disrupt efficient movement within nerve cells of RNA molecules. These RNA molecules direct protein assembly based on instructions carried in DNA. Correct transport of these RNAs permits proteins to be made in the right place at the right time.
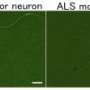
Working in motor neurons derived from patients with ALS, researchers demonstrated that each of three different TDP-43 mutations impaired delivery of RNA molecules to their final destination near the junction where a nerve and its target muscle meet. Without the RNA molecules, nerves cannot make proteins necessary to function normally and respond quickly when stimulated. Motor neurons govern movement, including breathing. Their death and deterioration is a hallmark of ALS.
The results also provide insight into how problems in RNA metabolism, including disturbances in RNA regulation and functioning, lead to ALS and other neurodegenerative diseases.
"Five years of tremendous progress in ALS genetics has revealed that RNA metabolism is a critical pathway that is impaired in this disease," said the study's corresponding author J. Paul Taylor, M.D., Ph.D., a member of the St. Jude Department of Developmental Neurobiology. "But RNA metabolism is a complex process that involves multiple steps that are carried out in different parts of the cell. This study provides a more refined understanding of how ALS-causing mutations impair RNA metabolism so we know what needs fixing therapeutically."
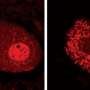
TDP-43 belongs to a family of proteins that bind to RNA and regulate its function. Normally TDP-43 is stored in the cell's command center, the nucleus. There the protein prepares DNA for translation into the proteins that do the work of cells and shuttles the resulting RNA, called mRNA, from the nucleus to the cytoplasm, the cell's liquid center. While clumps of TDP-43 were known to accumulate in the cytoplasm of the motor neurons of patients with ALS and other neurodegenerative diseases, the protein's function there was unknown.
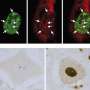
This study provides an answer. The work was done in motor neurons from the fruit fly Drosophila melanogaster, mouse brain cells and human motor neurons produced by reprogramming cells from ALS patients with three different TDP-43 mutations. Co-first author Nael Alami, Ph.D., a postdoctoral fellow in Taylor's laboratory, developed a florescent RNA beacon that let investigators track movement of RNA molecules in living cells.
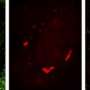
Researchers demonstrated that TDP-43 is part of a molecule called an RNA transport granule. These granules are responsible for moving mRNA efficiently to the end of the axon where the molecule is translated into a protein. For this study, scientists used Neurofilament-L (NEFL) mRNA, which is known to bind TDP-43.
In human motor neurons growing in the laboratory, investigators found that transport granules with mutant TDP-43 were more likely than granules with unaltered TDP-43 to stall en route to the nerve ending and sometimes reverse direction. The defect in the human ALS motor neurons was apparent after the first week.
Evidence from mice suggests TDP-43 mutations selectively rather than globally disrupt movement in nerve cells. The mutations did not affect movement of another cell structure, the mitochondria, along the axon where mRNA movement was impaired.
"We know neurodegenerative disorders, including Parkinson's and Alzheimer's diseases, seem to share a common mechanism," Alami said. "We plan to use our finding from this study to look for similar defects in those diseases."