Scientists uncover trigger for most common form of intellectual disability and autism

A new study led by Weill Cornell Medical College scientists shows that the most common genetic form of mental retardation and autism occurs because of a mechanism that shuts off the gene associated with the disease. The findings, published today in Science, also show that a drug that blocks this silencing mechanism can prevent fragile X syndrome – suggesting similar therapy is possible for 20 other diseases that range from mental retardation to multisystem failure.
Fragile X syndrome occurs mostly in boys, causing intellectual disability as well as telltale physical, behavioral and emotional traits. While researchers have known for more than two decades that the culprit behind the disease is an unusual mutation characterized by the excess repetition of a particular segment of the genetic code, they weren't sure why the presence of a large number of these repetitions – 200 or more – sets the disease process in motion.
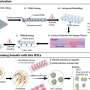
Using stem cells from donated human embryos that tested positive for fragile X syndrome, the scientists discovered that early on in fetal development, messenger RNA—a template for protein production—begins sticking itself onto the fragile X syndrome gene's DNA. This binding appears to gum up the gene, making it inactive and unable to produce a protein crucial to the transmission of signals between brain cells.
"Until 11 weeks of gestation, the fragile X syndrome gene is active – it produces its messenger RNA and protein normally. Then, all of a sudden it turns off, and stays off for the rest of the patient's lifetime, causing fragile X syndrome. But scientists have not understood why this gene gets shut off," says senior author Dr. Samie Jaffrey, a professor of pharmacology at Weill Cornell Medical College. "We discovered that the messenger RNA can jam up one strand of the gene's DNA, shutting down the gene—which was not known before.
"This is new biology—an interaction between the RNA and the DNA of the fragile X syndrome gene causes disease," Dr. Jaffrey says. "We are coming to understand that RNAs are powerful molecules that can regulate gene expression, but this mechanism is completely novel—and very exciting."
The malfunction occurs suddenly—before the end of the first trimester in humans and after 50 days in laboratory embryonic stem cells. At that point, the messenger RNA produced by the fragile X syndrome gene makes what the researchers call an RNA-DNA duplex—a particular arrangement of molecules in which the messenger RNA is stuck onto its DNA complement. (DNA produces two complementary strands of the genetic code responsible for human development and function. The four nucleic acids in the genomic code—A, C, G, T—have specific complements. In the case of fragile X syndrome, the repeat sequence in question is CGG. Therefore, RNA binds to its GCC complement on one strand of DNA.)
The RNA-DNA duplex then shuts down production of the fragile X syndrome gene, causing the loss of a protein needed for communication between brain cells. The gene then remains inactive for life. A normal fragile X gene—one with fewer than 200 CGG repeats—stays active in a person without the disorder, and produces the necessary protein. However, the mutant fragile X gene contains more than 200 CGG repeats, resulting in fragile X syndrome. Fragile X occurs in about 1 in 4,000 males and 1 in 8,000 females.
"Because the fragile X syndrome mutation is a repeat sequence, it is very easy for just a small portion of this sequence in the messenger RNA to find a matching repeat sequence on the DNA," Dr. Jaffrey says. "This is a unique feature of repeat sequences. When there are 200 or more repeats, the RNA-DNA interaction locks into place."
Hope for treatment – and other disorders
Dr. Jaffrey and his team, which includes researchers from The Scripps Research Institute in Florida and Albert Einstein College of Medicine in the Bronx, sought to find out why the disease is switched on when the CGG repeat is present in 200 to as many as 1,000 copies.
"Utilizing traditional ways to solve this puzzle has been impossible," he says. "Human fragile X syndrome genes introduced into mice and cells in the laboratory never turn off, no matter how many CGG repeats the genes have."
So the scientists turned to human embryonic stem cells. Co-authors Dr. Zev Rosenwaks, director and physician-in-chief of the Ronald O. Perelman and Claudia Cohen Center for Reproductive Medicine and director of the Stem Cell Derivation Laboratory of Weill Cornell Medical College, and Dr. Nikica Zaninovic, assistant professor of reproductive medicine, generated stem cell lines from donated embryos that tested positive for fragile X syndrome.
"These stem cells were critical to the success of this research, because they alone allowed us to mimic what happens to the fragile X gene during embryonic development," says Dr. Dilek Colak, a postdoctoral scientist in Dr. Jaffrey's laboratory and the first author of the study.
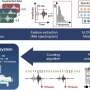
The stem cells were coaxed to become brain neurons, and at about 50 days, they differentiated in the same way that an embryo's brain is developing at 11-plus weeks when the fragile X syndrome gene is switched off.
The researchers then used a drug developed by co-author Dr. Matthew Disney of the Scripps Research Institute that binds to CGG in the fragile X gene's RNA before and after the 50-day switch. Strikingly, the gene never stopped producing its beneficial protein.
That suggests a potential prevention or treatment strategy for fragile X syndrome, Dr. Jaffrey says. "If a pregnant woman is told that her fetus carries the genetic mutation causing fragile X syndrome, we could potentially intervene and give the drug during gestation. This may delay or prevent the silencing of the fragile X gene, which could potentially significantly improve the outcome of these patients," he says.
The researchers are now looking for similar RNA-DNA duplexes in other trinucleotide repeat diseases, including Huntington's disease (a degenerative brain disease), myotonic dystrophy 1 and 2 (a multisystem progressive disease), Friedrich's ataxia (a progressive nervous system disorder), Jacobsen syndrome (an intellectual disorder), and familial amyotrophic lateral sclerosis (a motor neuron disease), among others.
"This completely new mechanism by which RNAs can direct gene silencing may be involved in a lot of other diseases," Dr. Jaffrey says. "Our hope is that we can find drugs that interfere with this new type of disease process."