Computational methods for studying gene and protein function
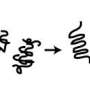
 interacts with protein X (orange) via a coiled coil interaction, the polyQ adopts a coiled coil conformation, which extends the preceding coiled coil (blue). The polyP region (green) cannot adopt a coiled coil conformation, effectively capping the interaction-dependent conformational change of the polyQ region.")
Miguel Andrade uses computational methods for studying gene and protein function with an emphasis on molecules related to human disease.
With the vast amount of data generated in modern molecular biology research, powerful methods are needed to understand and make use of the results. For instance, new techniques let researchers model protein interactions, predict how the transcription of genes is regulated, and visualize how genes are expressed, both in health and disease. Professor Miguel Andrade's work spans all these areas, plus data and text mining of the biomedical literature. These strands of research are connected by the computational tools developed and applied by his group to make sense of large data sets from biological experiments.
One such tool is HIPPIE, a database that integrates protein-protein interaction data. Combining information from genomic, phylogenetic and functional sources, for example, Andrade has shown that stretches of polyglutamine (polyQ) in proteins modulate interactions with other proteins (see Figure). Abnormal expansion of polyQ can result in pathological protein aggregation, which may contribute to the disease mechanism in various neurodegenerative conditions, such as Huntington's disease or spinocerebellar ataxia.
One of the main goals of Andrade's work is the prediction of protein and gene function by integrating heterogeneous data. An example of this is his recent observation that microRNA targets and functions can be better predicted using data about the targets of transcriptional suppressor proteins. This finding has been applied to the neural repressor protein REST to identify candidate miRNAs that could act as suppressors of brain cancer.
Another aspect of Andrade's work concerns the development of methods for data and text mining of the biomedical literature, for example abstracts of scientific papers in the PubMed database. Fast prioritization of hundreds of thousands of PubMed records according to user-defined topics in a matter of seconds allows efficient exploration of the biomedical corpus, and can be used to sort genes and chemicals according to their relevance to a topic. Professor Miguel Andrade's group has also used PubMed to study economic, linguistic and scientific trends, and to find appropriate reviewers for manuscripts.
Professor Christof Niehrs, IMB's Founding Director, says the recruitment of Miguel Andrade is a boon for IMB as well as for the wider biology community in Mainz. "Miguel Andrade's computational work ideally complements the wet lab expertise we already have at IMB. As a leader in bioinformatics, Miguel is a pivotal asset for the life sciences in Mainz."