Credit: © Mary Ann Liebert, Inc., publishers
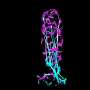
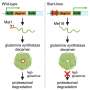
To make up for insufficient amounts of SMN protein, the cause of the inherited neuromuscular disease spinal muscular atrophy (SMA), researchers have successfully delivered a replacement SMN1 gene directly to the spinal cords of animal models of SMA. A new study demonstrating that enough copies of the SMN1 gene can be delivered to the spinal cord motor neurons to extend the survival of the treated animals is published in Human Gene Therapy.
Marco Passini and coauthors from Genzyme (Framingham, MA), University of California San Francisco, Emory University School of Medicine (Atlanta, GA), and Georgetown University Medical Center (Washington, DC) used an adeno-associated viral vector as the delivery vehicle to transport copies of the SMN1 gene into motor neurons in the spinal cord via intrathecal delivery. They report on the effectiveness of restoring the levels of functional SMN protein in normal pig and non-human primate SMA models that would predict efficacy based on gene transfer with the same vector in an authentic mouse model of SMA in the article "Translational Fidelity of Intrathecal Delivery of Self-Complementary AAV9–Survival Motor Neuron 1 for Spinal Muscular Atrophy."
"This is a very promising and thorough set of preclinical studies that supports rapid translation to the clinic," says James M. Wilson, MD, PhD, Editor-in-Chief of Human Gene Therapy, and Director of the Gene Therapy Program, Department of Pathology and Laboratory Medicine, University of Pennsylvania Perelman School of Medicine, Philadelphia.
More information: The article is available free on the Human Gene Therapy website.
Journal information: Human Gene Therapy
Provided by Mary Ann Liebert, Inc