Fatal cellular malfunction identified in Huntington's disease

Researchers believe they have learned how mutations in the gene that causes Huntington's disease kill brain cells, a finding that could open new opportunities for treating the fatal disorder. Scientists first linked the gene to the inherited disease more than 20 years ago.
Huntington's disease affects five to seven people out of every 100,000. Symptoms, which typically begin in middle age, include involuntary jerking movements, disrupted coordination and cognitive problems such as dementia. Drugs cannot slow or stop the progressive decline caused by the disorder, which leaves patients unable to walk, talk or eat.
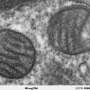
Lead author Hiroko Yano, PhD, of Washington University School of Medicine in St. Louis, found in mice and in mouse brain cell cultures that the disease impairs the transfer of proteins to energy-making factories inside brain cells. The factories, known as mitochondria, need these proteins to maintain their function. When disruption of the supply line disables the mitochondria, brain cells die.
"We showed the problem could be fixed by making cells overproduce the proteins that make this transfer possible," said Yano, assistant professor of neurological surgery, neurology and genetics. "We don't know if this will work in humans, but it's exciting to have a solid new lead on how this condition kills brain cells."
The findings are available online in Nature Neuroscience.
Huntington's disease is caused by a defect in the huntingtin gene, which makes the huntingtin protein. Life expectancy after initial onset is about 20 years.
Scientists have known for some time that the mutated form of the huntingtin protein impairs mitochondria and that this disruption kills brain cells. But they have had difficulty understanding specifically how the gene harms the mitochondria.
For the new study, Yano and collaborators at the University of Pittsburgh worked with mice that were genetically modified to simulate the early stages of the disorder.
Yano and her colleagues found that the mutated huntingtin protein binds to a group of proteins called TIM23. This protein complex normally helps transfer essential proteins and other supplies to the mitochondria. The researchers discovered that the mutated huntingtin protein impairs that process.
The problem seems to be specific to brain cells early in the disease. At the same point in the disease process, the scientists found no evidence of impairment in liver cells, which also produce the mutated huntingtin protein.
The researchers speculated that brain cells might be particularly reliant on their mitochondria to power the production and recycling of the chemical signals they use to transmit information. This reliance could make the cells vulnerable to disruption of the mitochondria.
Other neurodegenerative conditions, including Alzheimer's disease and amyotrophic lateral sclerosis, also known as Lou Gehrig's disease, have been linked to problems with mitochondria. Scientists may be able to build upon these new findings to better understand these disorders.
More information: Yano H, Baranov SV, Baranova OV, Kim J, Pan Y, Yablonska S, Carlisle DL, Ferrante RJ, Kim AH, Friedlander RM. Inhibition of mitochondrial protein import by mutant huntingtin. Nature Neuroscience, published online May 18, 2014.