Forcing chromosomes into loops may switch off sickle cell disease
Scientists have altered key biological events in red blood cells, causing the cells to produce a form of hemoglobin normally absent after the newborn period. Because this hemoglobin is not affected by the inherited gene mutation that causes sickle cell disease, the cell culture findings may give rise to a new therapy for the debilitating blood disorder.
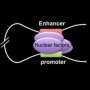
The novel approach uses protein-engineering techniques to force chromatin fiber, the substance of chromosomes, into looped structures that contact DNA at specific sites to preferentially activate genes that regulate hemoglobin. "We have demonstrated a novel way to reprogram gene expression in blood-forming cells," said study leader Gerd A. Blobel, M.D., Ph.D., who holds the Frank E. Weise III Endowed Chair in Pediatric Hematology at The Children's Hospital of Philadelphia. "If we can translate this approach into the clinic, this may become a new treatment for patients with sickle-cell disease."
Blobel and colleagues, including Wulan Deng, Ph.D., formerly a member of the Blobel laboratory, and current lab member Jeremy W. Rupon, M.D., Ph.D., published their findings online today in Cell.
Key to the researcher's strategy is a developmental transition that normally occurs in the blood of newborns. A biological switch regulates a changeover from fetal hemoglobin to adult hemoglobin as it begins to silence the genes that produce fetal hemoglobin. This has major consequences for patients with the mutation that causes sickle cell disease (SCD).
Fetal hemoglobin is not affected by this mutation. But as adult hemoglobin starts to predominate, patients with the SCD mutation begin to experience painful, sometimes life-threatening disease symptoms as misshapen red blood cells disrupt normal circulation, clog blood vessels and damage organs.
Hematologists have long known that sickle cell patients with elevated levels of fetal hemoglobin compared to adult hemoglobin have a milder form of the disease. "This observation has been a major driver in the field to understand the molecular basis of the mechanisms that control the biological switch, with the ultimate goal to reverse it," said Blobel.
In previous research, Blobel's team used bioengineering techniques to adapt zinc-finger proteins to latch onto specific DNA sites far apart on a chromosome. The chromatin loop that results transmits regulatory signals for specific genes.
In their current work, the scientists custom-designed zinc fingers to flip the biological switch in blood-forming cells, reactivating the genes expressing fetal hemoglobin at the expense of the genes expressing adult hemoglobin. The researchers achieved these results in cultured blood cells from adult mice and adult humans.
The next step, said Blobel, is to apply this proof-of-concept technique to preclinical models, by testing the approach in animals genetically engineered to have manifestations of SCD similar to that found in human patients. If this strategy corrects the disease in animals, it may set the stage to move to human trials.
In principle, added Blobel, the forced chromatin looping approach could also be applied to other hemoglobin-related disorders, such as certain forms of thalassemia in which elevated fetal hemoglobin levels might be beneficial.
More information: Wulan Deng et al, "Reactivation of developmentally silenced globin genes by forced chromatin looping," Cell, published online Aug. 14, 2014.