A new player in lipid metabolism discovered
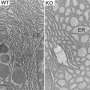
 became obese, experimental mice (AKO, adipocyte-specific Sel1L knockout mice) lacking the SEL1L gene in their fat cells stayed lean. Credit: Haibo Sha")
(Medical Xpress)—Specially engineered mice that lacked a particular gene did not gain weight when fed a typical high-fat, obesity-inducing Western diet. Yet, these mice ate the same amount as their normal counterparts that became obese.
The mice were engineered with fat cells that lacked a gene called SEL1L, known to be involved in the clearance of misfolded proteins in the cell's protein making machinery called the endoplasmic reticulum (ER).
When misfolded proteins are not cleared but accumulate, they destroy the cell and contribute to such diseases as mad cow disease, Type 1 diabetes and cystic fibrosis.
"The million-dollar question is why don't these mice gain weight? Is this related to its inability to clear misfolded proteins in the ER?" said Ling Qi, associate professor of molecular and biochemical nutrition and senior author of the study published online July 24 in Cell Metabolism. Haibo Sha, a research associate in Qi's lab, is the paper's lead author.
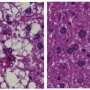
Interestingly, the experimental mice developed a host of other problems, including postprandial hypertriglyceridemia, which is characterized by high levels of triglycerides in humans and mice (after a meal) that can lead to heart attacks and strokes if untreated, and fatty livers.
"Although we are yet to find out whether these conditions contribute to the lean phenotype, we found that there was a lipid partitioning defect in the mice lacking SEL1L in fat cells, where fat cells cannot store fat [lipids], and consequently fat goes to the liver. During the investigation of possible underlying mechanisms, we discovered a novel function for SEL1L as a regulator of lipid metabolism," said Qi.
"We were very excited to find that SEL1L is required for the intracellular trafficking of an enzyme called lipoprotein lipase (LPL), acting as a chaperone," said Sha. "Using several tissue-specific knockout mouse models, we showed that this is a general phenomenon," Sha added.
Without LPL, lipids remain in the circulation; fat and muscle cells cannot absorb fat molecules for storage and energy combustion, respectively. Many humans have LPL mutations and develop postprandial hypertriglyceridemia similar to conditions found in fat cell-specific SEL1L-deficient mice, said Qi.
Future work will investigate the role of SEL1L in human patients carrying LPL mutations and determine why fat cell-specific SEL1L-deficient mice remain lean under Western diets, said Sha.